

Секвенування всього екзому людини

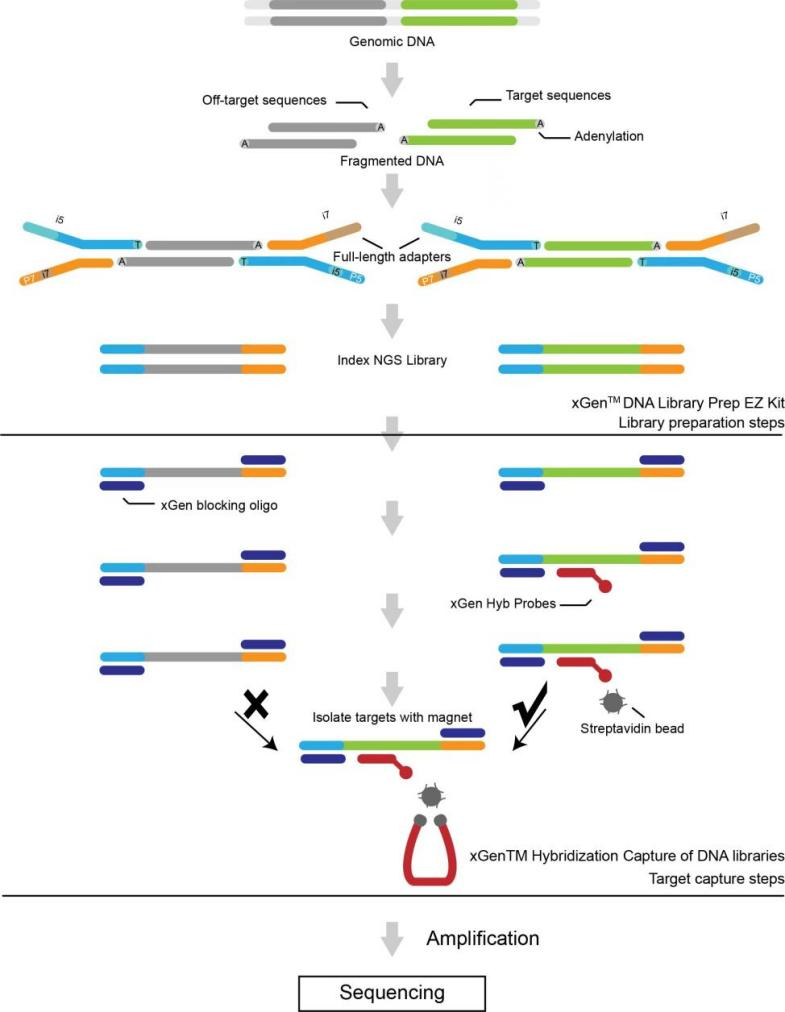
Особливості сервісу
● Доступні дві панелі екзомів на основі цільового збагачення зондами: Sure Select Human All Exon v6 (Agilent) і xGen Exome Hybridization Panel v2 (IDT).
● Секвенування на Illumina NovaSeq.
● Біоінформаційний конвеєр, спрямований на аналіз захворювання чи пухлини.
Переваги сервісу
●Націлена на область кодування білка: шляхом захоплення та секвенування ділянок, що кодують білок, hWES використовується для виявлення варіантів, пов’язаних зі структурою білка.
●Рентабельність:hWES дає приблизно 85% мутацій, пов’язаних із захворюваннями людини, з 1% геному людини.
●Висока точність: Завдяки високій глибині секвенування hWES полегшує виявлення як поширених варіантів, так і рідкісних варіантів із частотою нижче 1%.
●Суворий контроль якості: Ми впроваджуємо п’ять основних контрольних точок на всіх етапах, від підготовки зразків і бібліотеки до секвенування та біоінформатики. Такий ретельний моніторинг забезпечує стабільно високу якість результатів.
●Комплексний аналіз біоінформатики: наш набір виходить за рамки ідентифікації варіацій еталонного геному, оскільки він включає розширений аналіз, розроблений спеціально для вирішення дослідницьких питань, пов’язаних із генетичними аспектами захворювань або аналізом пухлин.
●Підтримка після продажу:Наше зобов’язання поширюється на період після завершення проекту з 3-місячним періодом післяпродажного обслуговування. Протягом цього часу ми пропонуємо подальше спостереження за проектом, допомогу з усунення несправностей і сеанси запитань і відповідей, щоб відповісти на будь-які запитання, пов’язані з результатами.
Зразок специфікації
| Стратегія захоплення екзонів | Стратегія секвенування | Рекомендований вихід даних |
| Sure Select Human All Exon v6 (Agilent) або xGen Exome Hybridization Panel v2 (IDT)
| Illumina NovaSeq PE150 | 5-10 Гб Для менделівських розладів/рідкісних захворювань: > 50x Для зразків пухлин: ≥ 100x |
Зразок вимог
| Тип зразка
| Сума(Qubit®)
| Концентрація | Обсяг
| Чистота (NanoDrop™) |
|
Геномна ДНК
| ≥ 50 нг | ≥ 6 нг/мкл | ≥ 15 мкл | OD260/280=1,8-2,0 без деградації, без забруднення
|
Біоінформатика
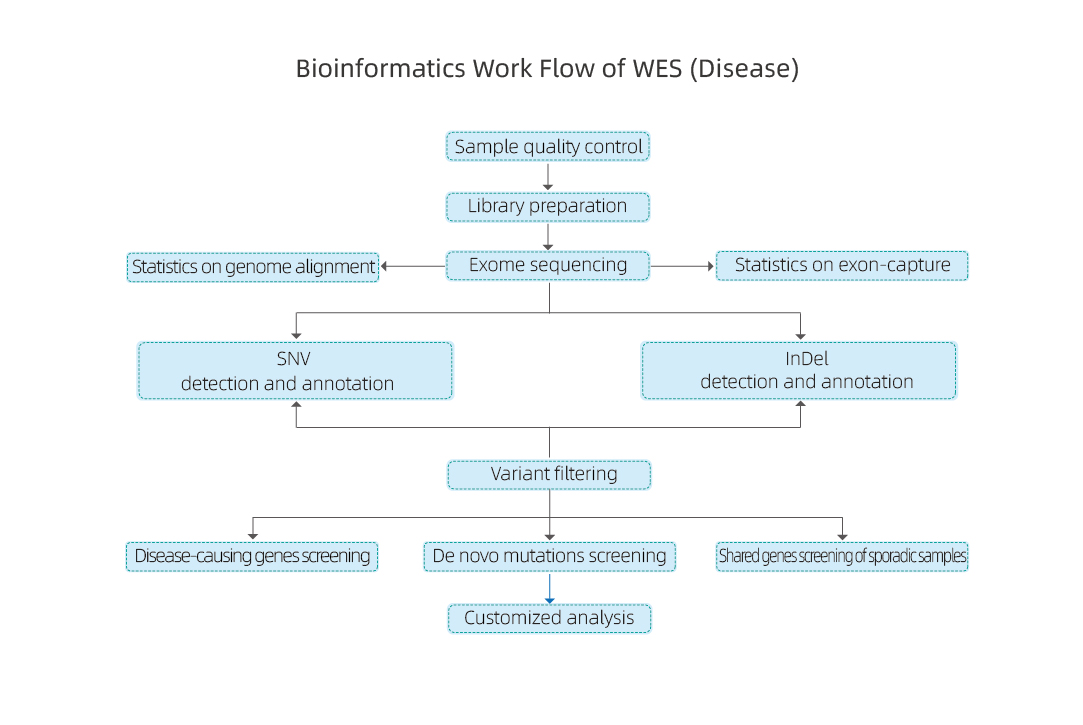
Біоінформаційний аналіз зразків hWES-захворювання включає:
● КЯ даних секвенування
● Вирівнювання еталонного геному
● Ідентифікація SNP та InDels
● Функціональна анотація SNP та InDels
Біоінформаційний аналіз зразків пухлин включає:
● КЯ даних секвенування
● Вирівнювання еталонного геному
● Ідентифікація SNP, InDels і соматичних варіацій
● Ідентифікація варіантів зародкової лінії
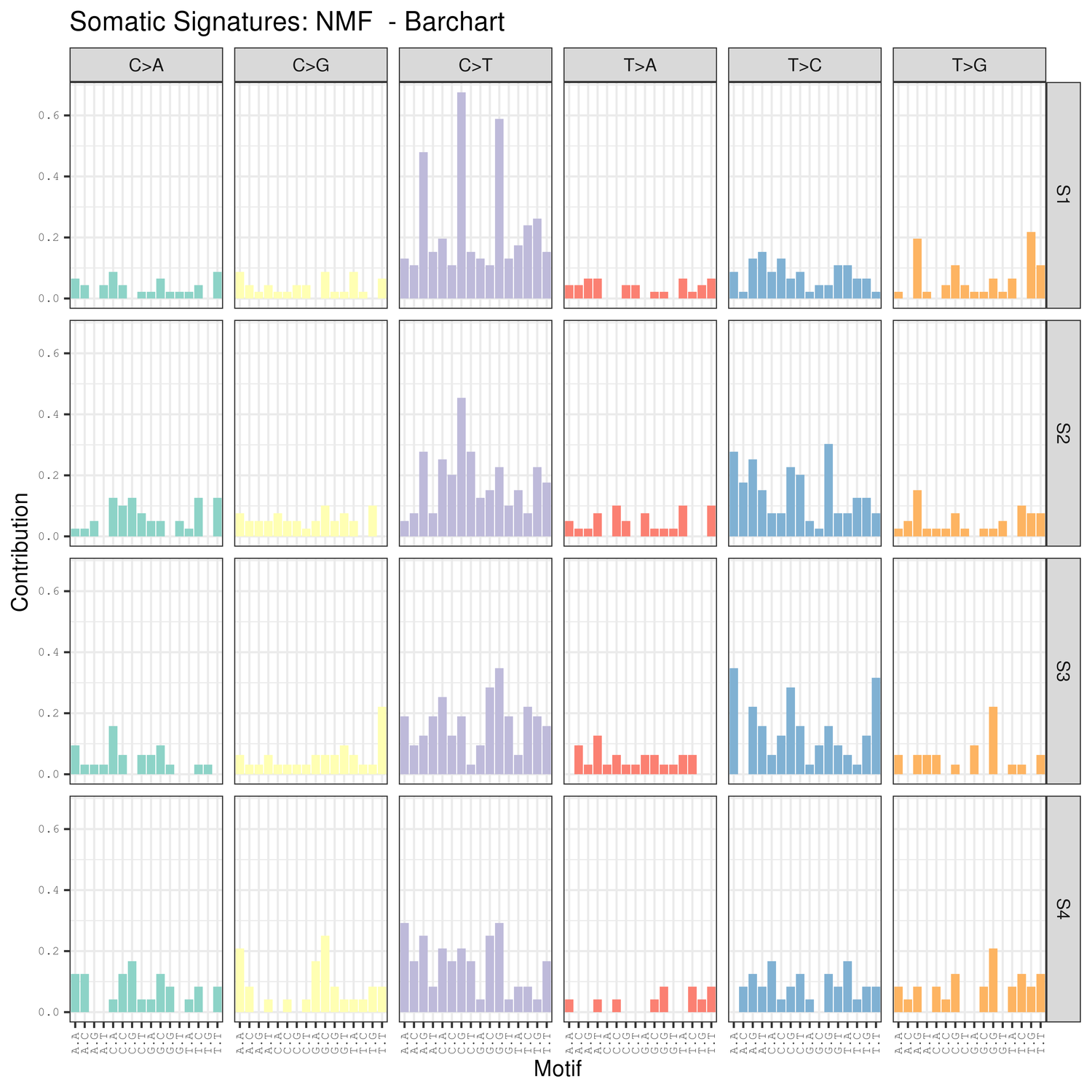
● Аналіз сигнатур мутації
● Ідентифікація генів приводу на основі мутацій посилення функції
● Анотація мутації на рівні лікарської чутливості
● Аналіз гетерогенності – розрахунок чистоти та плоїдності
Потік роботи служби
Доставка зразків

екстракція ДНК

Будівництво бібліотеки

Секвенування

Аналіз даних
Доставка даних

Післяпродажне обслуговування
Контроль якості даних – статистика захоплення Exome

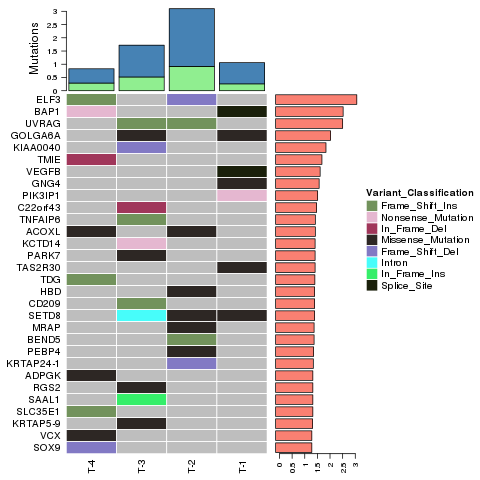
Ідентифікація варіантів – InDels

Розширений аналіз: ідентифікація та розподіл шкідливих SNP/InDels – графік Circos

Аналіз пухлини: ідентифікація та розподіл соматичних мутацій – графік Circos

Аналіз пухлин: клонові лінії






