

การวิเคราะห์แบบแยกเป็นจำนวนมาก

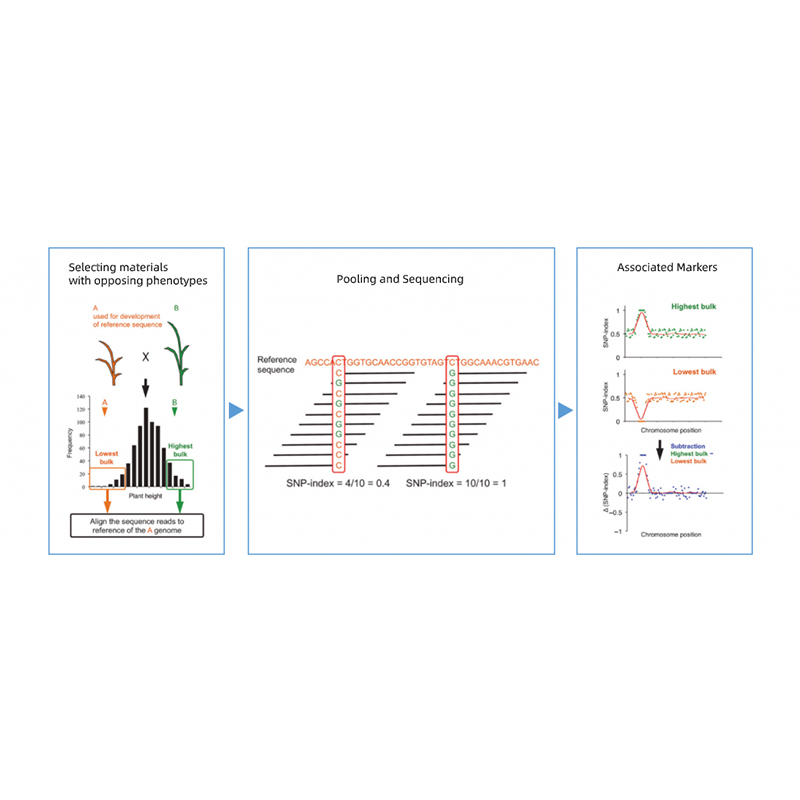
ข้อได้เปรียบด้านการบริการ
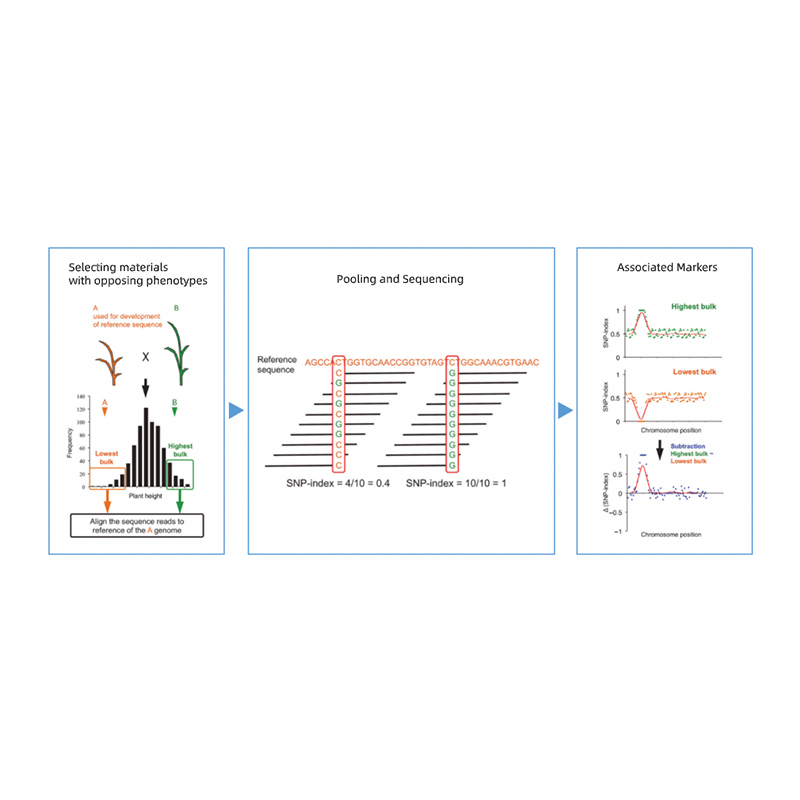
Takagi et al., The Plant Journal, 2013
●การแปลที่แม่นยำ: การผสม bulks กับ 30+30 ถึง 200+200 คนเพื่อลดเสียงรบกวนพื้นหลัง การทำนายภูมิภาคผู้สมัครที่ไม่ระบุชื่อที่ไม่ระบุชื่อ
●การวิเคราะห์ที่ครอบคลุม: คำอธิบายประกอบฟังก์ชั่นการทำงานของผู้สมัครในเชิงลึกรวมถึง NR, SwissProt, GO, KEGG, COG, KOG, ฯลฯ
●เวลาตอบสนองที่เร็วขึ้น: การแปลยีนอย่างรวดเร็วภายใน 45 วันทำการ
●ประสบการณ์ที่กว้างขวาง: BMK มีส่วนร่วมในการแปลหลายพันลักษณะครอบคลุมสายพันธุ์ที่หลากหลายเช่นพืชผลผลิตภัณฑ์น้ำป่าไม้ดอกไม้ผลไม้ ฯลฯ
ข้อกำหนดบริการ
ประชากร:
การแยกลูกหลานของผู้ปกครองด้วยฟีโนไทป์ที่ต่อต้าน
เช่นลูกหลาน F2, backcrossing (BC), recombinant สายพันธุ์ (RIL)
สระผสม
สำหรับลักษณะเชิงคุณภาพ: 30 ถึง 50 คน (ขั้นต่ำ 20)/กลุ่ม
สำหรับ tratis เชิงปริมาณ: 5% ถึง 10% บุคคลที่มีฟีโนไทป์ที่รุนแรงในประชากรทั้งหมด (ขั้นต่ำ 30+30)
ความลึกการเรียงลำดับที่แนะนำ
อย่างน้อย 20x/parent และ 1x/rofspring บุคคล (เช่นสำหรับการผสมของลูกหลานที่ 30+30 แต่ละรายการความลึกการเรียงลำดับจะเป็น 30x ต่อเป็นกลุ่ม)
การวิเคราะห์ทางชีวสารสนเทศศาสตร์
●การ resequencing จีโนมทั้งหมด
●การประมวลผลข้อมูล
●การโทร SNP/Indel
●การคัดกรองภูมิภาคผู้สมัคร
●คำอธิบายประกอบการทำงานของยีนของผู้สมัคร
ข้อกำหนดตัวอย่างและการจัดส่ง
ข้อกำหนดตัวอย่าง:
นิวคลีโอไทด์:
| ตัวอย่าง gdna | ตัวอย่างเนื้อเยื่อ |
| ความเข้มข้น: ≥30 ng/μl | พืช: 1-2 กรัม |
| จำนวน: ≥2μg (Volumn ≥15μl) | สัตว์: 0.5-1 กรัม |
| ความบริสุทธิ์: OD260/280 = 1.6-2.5 | เลือดทั้งหมด: 1.5 มล. |
งานบริการ

การออกแบบการทดลอง

การส่งตัวอย่าง

การสกัด RNA

การก่อสร้างห้องสมุด

การเรียงลำดับ

การวิเคราะห์ข้อมูล

บริการหลังการขาย
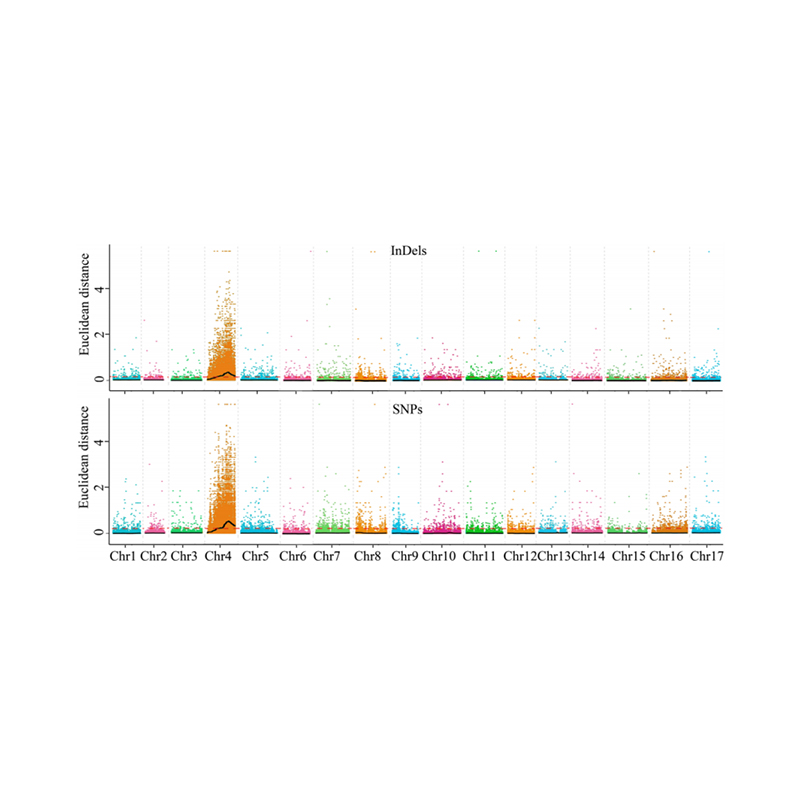
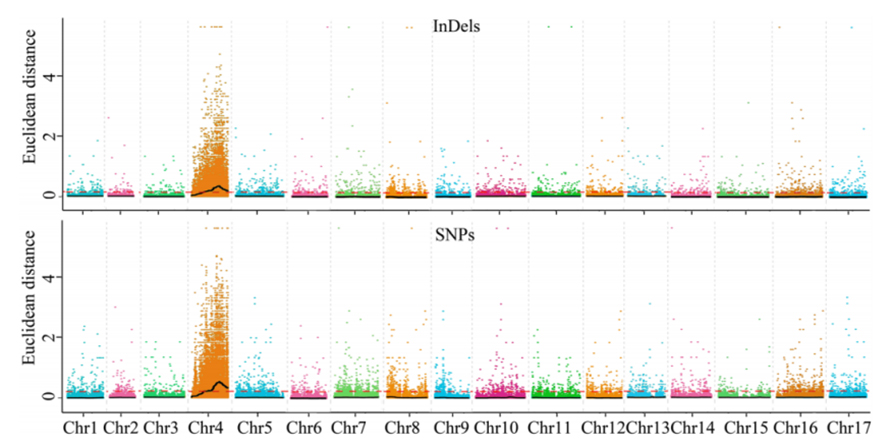
1. ฐานการวิเคราะห์การเชื่อมโยงกับระยะทางยุคลิด (ED) เพื่อระบุภูมิภาคผู้สมัคร ในรูปต่อไปนี้
แกน x: หมายเลขโครโมโซม; แต่ละจุดแสดงค่า ED ของ SNP เส้นสีดำสอดคล้องกับค่า ED ที่ติดตั้ง ค่า ED ที่สูงขึ้นหมายถึงความสัมพันธ์ที่สำคัญยิ่งขึ้นระหว่างไซต์และฟีโนไทป์ เส้นประสีแดงแสดงถึงเกณฑ์ของการเชื่อมโยงที่สำคัญ
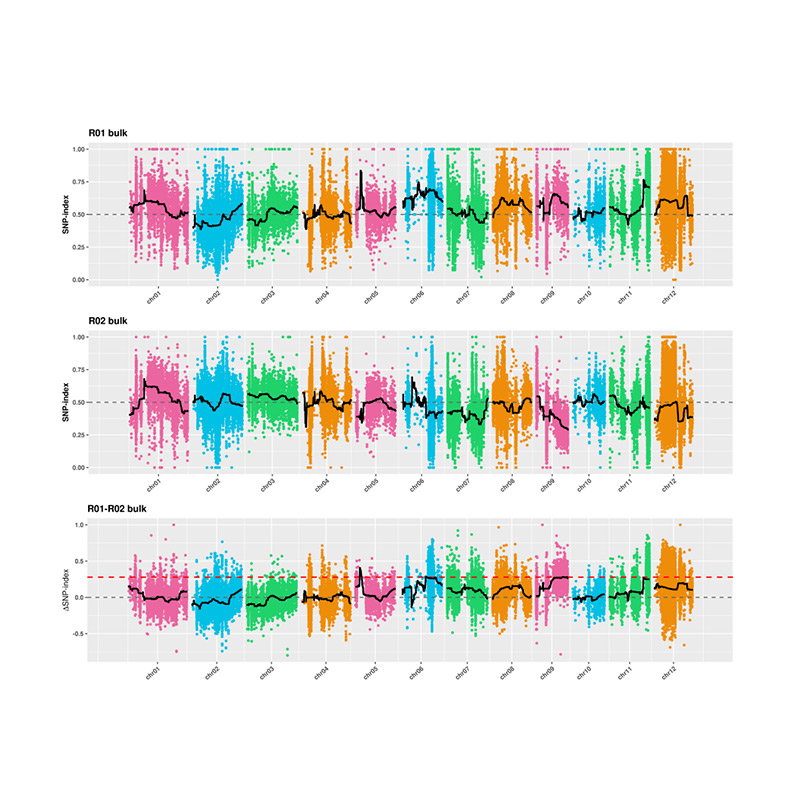
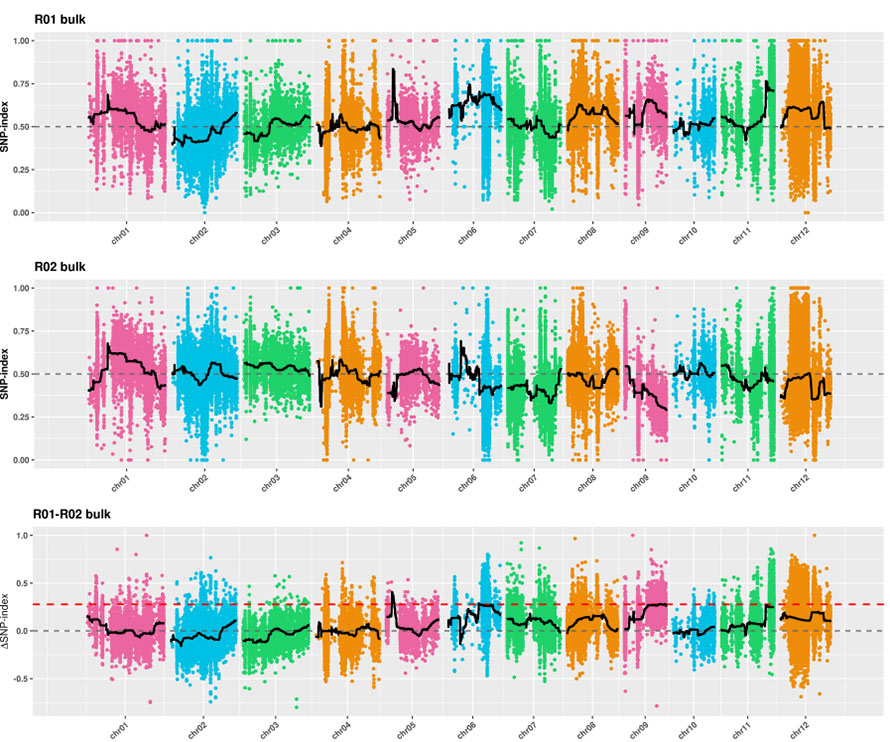
2. การวิเคราะห์การเชื่อมโยงไม่มีดัชนี SNP
แกน x: หมายเลขโครโมโซม; แต่ละจุดแสดงค่าดัชนี SNP เส้นสีดำหมายถึงค่าดัชนี SNP ที่ติดตั้ง ค่าที่ใหญ่ขึ้นคือยิ่งความสัมพันธ์มีความสำคัญมากขึ้นเท่านั้น
กรณี BMK
ลักษณะเชิงปริมาณที่สำคัญ-ผลการศึกษา FNL7.1 เข้ารหัสตัวอ่อนสายโปรตีนมากมายที่เกี่ยวข้องกับความยาวคอผลไม้ในแตงกวา
ที่ตีพิมพ์: วารสารเทคโนโลยีชีวภาพพืช, 2020
กลยุทธ์การเรียงลำดับ:
ผู้ปกครอง (JIN5-508, YN): จีโนมทั้งหมด resequencing สำหรับ 34 ×และ 20 ×
พูล DNA (50 คอยาวและ 50 คอสั้น): resequencing สำหรับ 61 ×และ 52 ×
ผลลัพธ์ที่สำคัญ
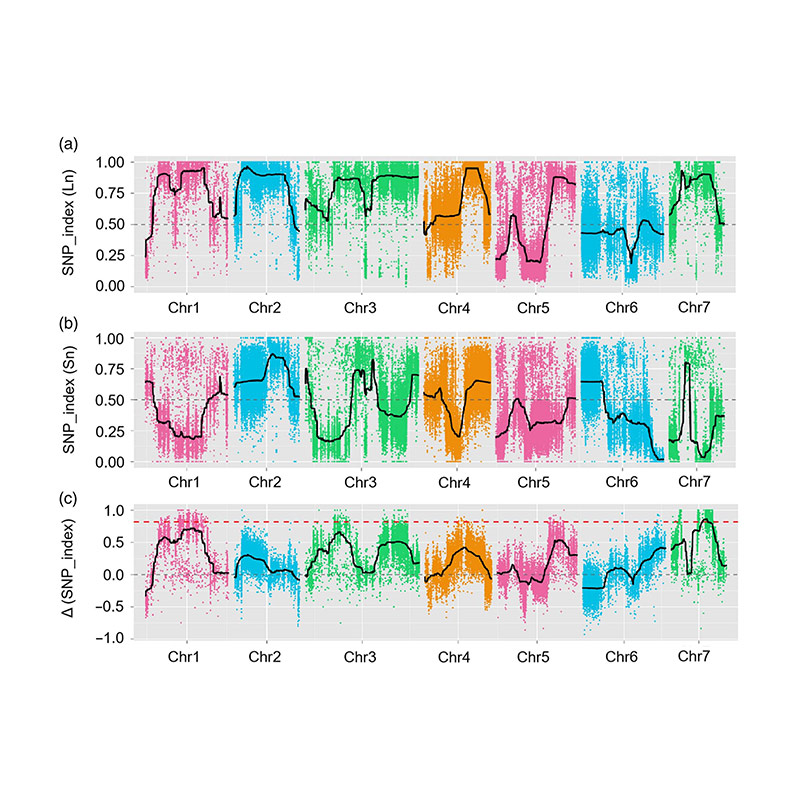
ในการศึกษานี้การแยกประชากร (F2 และ F2: 3) ถูกสร้างขึ้นโดยการข้ามสายแตงกวาคอยาว Jin5-508 และคอสั้นคอ สระดีเอ็นเอสองสระถูกสร้างขึ้นโดย 50 คนที่มีคอยาวมากและ 50 คนคอสั้นสุดขีด QTL เอฟเฟกต์ที่สำคัญถูกระบุในการวิเคราะห์ CHR07 โดยการวิเคราะห์ BSA และการทำแผนที่ QTL แบบดั้งเดิม ภูมิภาคของผู้สมัครถูก จำกัด ให้แคบลงโดยการทำแมปปริมาณการแสดงออกของยีนและการทดลองดัดแปลงพันธุกรรมซึ่งเผยให้เห็นยีนที่สำคัญในการควบคุมความยาวคอ, CSFNL7.1 นอกจากนี้ความหลากหลายในภูมิภาคโปรโมเตอร์ CSFNL7.1 พบว่าเกี่ยวข้องกับการแสดงออกที่สอดคล้องกัน การวิเคราะห์ทางสายวิวัฒนาการเพิ่มเติมชี้ให้เห็นว่า FNL7.1 locus มีแนวโน้มที่จะเกิดขึ้นจากอินเดีย
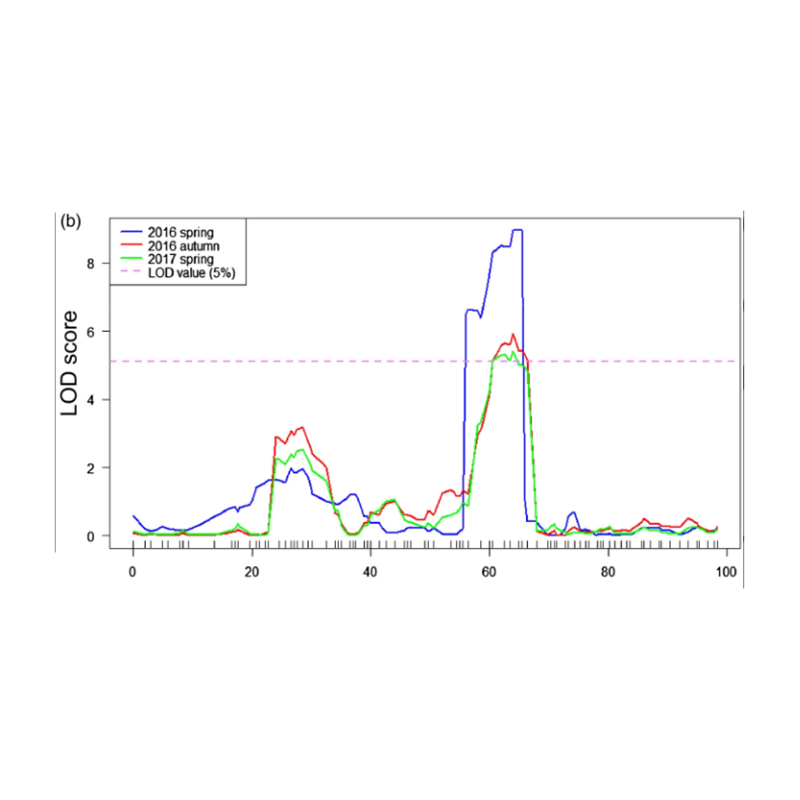
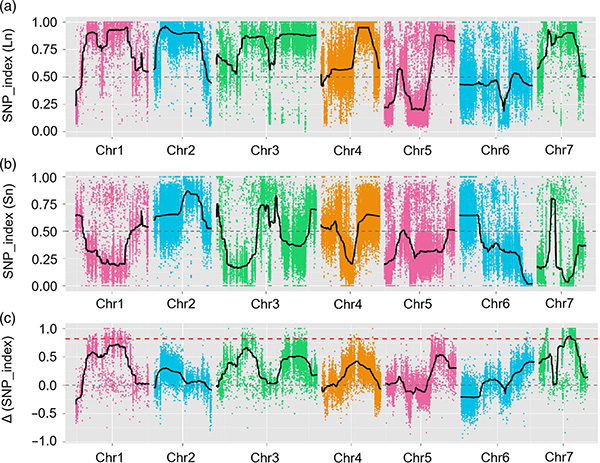 การแมป QTL ในการวิเคราะห์ BSA เพื่อระบุภูมิภาคผู้สมัครที่เกี่ยวข้องกับความยาวคอแตงกวา | 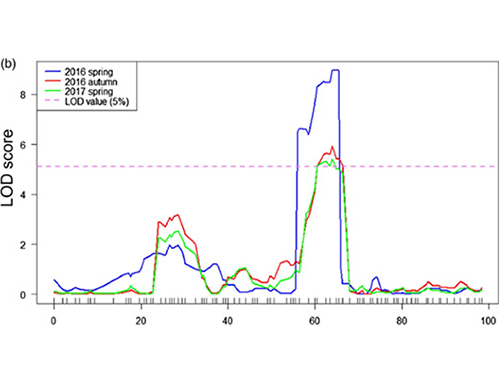 โปรไฟล์ LOD ของ qtl ความยาวคอแตงกวาที่ระบุไว้ใน Chr07 |
Xu, X. , et al. “ ลักษณะเชิงปริมาณที่สำคัญของ Locus FNL7.1 เข้ารหัสโปรตีน embryogenesis ปลายที่มีความยาวคอผลไม้ในแตงกวา” วารสารเทคโนโลยีชีวภาพพืช 18.7 (2020)



