

Jämförande genomik

Servicefördelar
●Omfattande expertis och publikationsregister: med ackumulerat har BMKGene slutfört över 90 jämförande genomikprojekt, med en kumulativ effektfaktor nådd 900.
●Omfattande bioinformatikanalys: analyspaketet innehåller de åtta vanligaste analyserna, vilket ger väldesignade siffror redo att publiceras och möjliggör en enkel tolkning av resultaten
●Mycket kompetent bioinformatikteam och kort analyscykel: med stor erfarenhet av jämförande genomikanalys, uppfyller BMKGenes team olika personliga analyskrav på kort handläggningstid
●Support efter försäljning:Vårt engagemang sträcker sig längre än att slutföra projekt med en 3-månaders serviceperiod efter försäljning. Under denna tid erbjuder vi projektuppföljning, felsökningshjälp och frågestunder för att ta itu med eventuella frågor relaterade till resultaten.
Servicespecifikationer
| Beräknad handläggningstid | Antal arter | Analyser |
| 30 arbetsdagar | 6 - 12 | Genfamiljklustring Genfamiljens expansion och sammandragning Filogenetisk trädkonstruktion Uppskattning av divergenstiden (fossil kalibrering krävs) LTR-insättningstid (för växter) Helgenomduplicering (för växter) Selektivt tryck Synteny analys |
Bioinformatikanalyser
● Genfamilj
● Fylogenetik
● Divergenstid
● Selektivt tryck
● Syntenyanalys
Exempel på krav och leverans
Exempelkrav:
Vävnad eller DNA för genomsekvensering och sammansättning
För vävnad
| Art | Vävnad | Undersökning | PacBio CCS |
| Djur | Visceral vävnad | 0,5 ~ 1 g | ≥ 3,5 g |
| Muskelvävnad | |||
| ≥ 5,0 g | |||
| ≥ 5,0 ml | |||
| Däggdjursblod | |||
| ≥ 0,5 ml | |||
| Fjäderfä/fiskblod | |||
| Plantera | Färskt blad | 1 ~ 2 g | ≥ 5,0 g |
| Kronblad/Stjälk | 1 ~ 2 g | ≥ 10,0 g | |
| Rot/Frö | 1 ~ 2 g | ≥ 20,0 g | |
| Celler | Odlad cell | - | ≥ 1 x 108 |
Data
Genomsekvensfiler (.fasta) och anteckningsfiler (.gff3) av närbesläktade arter
Service Arbetsflöde

Experimentdesign

Provleverans

Byggande av bibliotek

Sekvensering

Dataanalys

Service efter försäljning
*Demoresultat som visas här är alla från genom publicerade med Biomarker Technologies
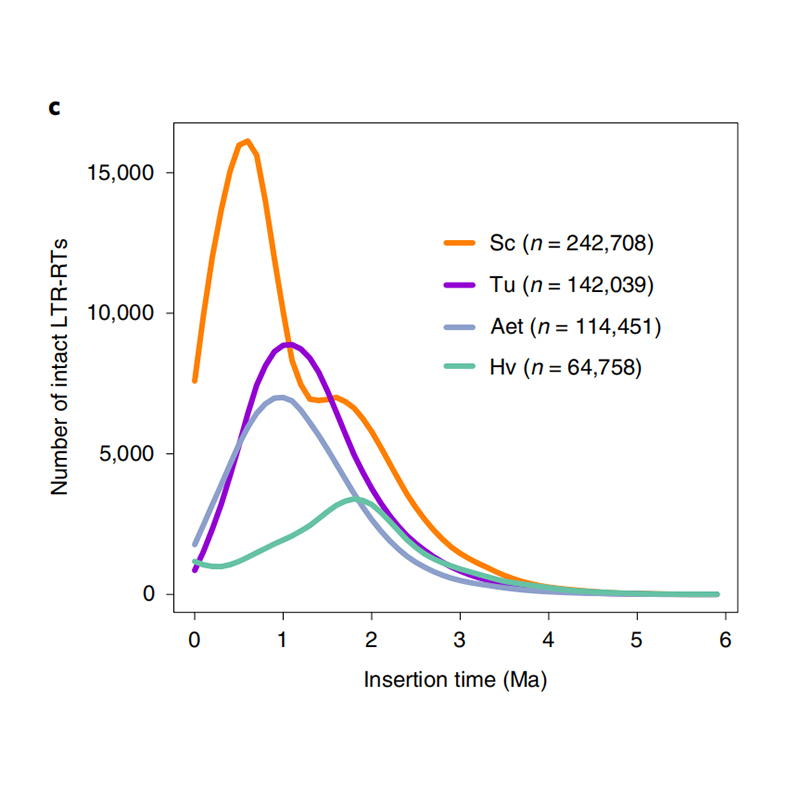
1.LTR-insättningstidsuppskattning: Figuren visade en unik bimodal fördelning i LTR-RTs insättningstider i Weining råggenom, jämfört med andra arter. Den senaste toppen dök upp för cirka 0,5 miljoner år sedan.
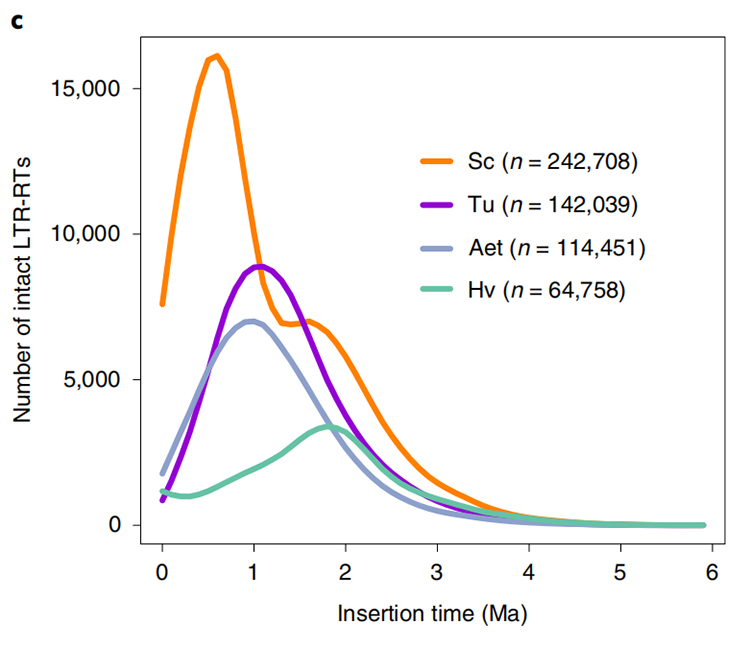
Li Guang et al.,Naturgenetik, 2021
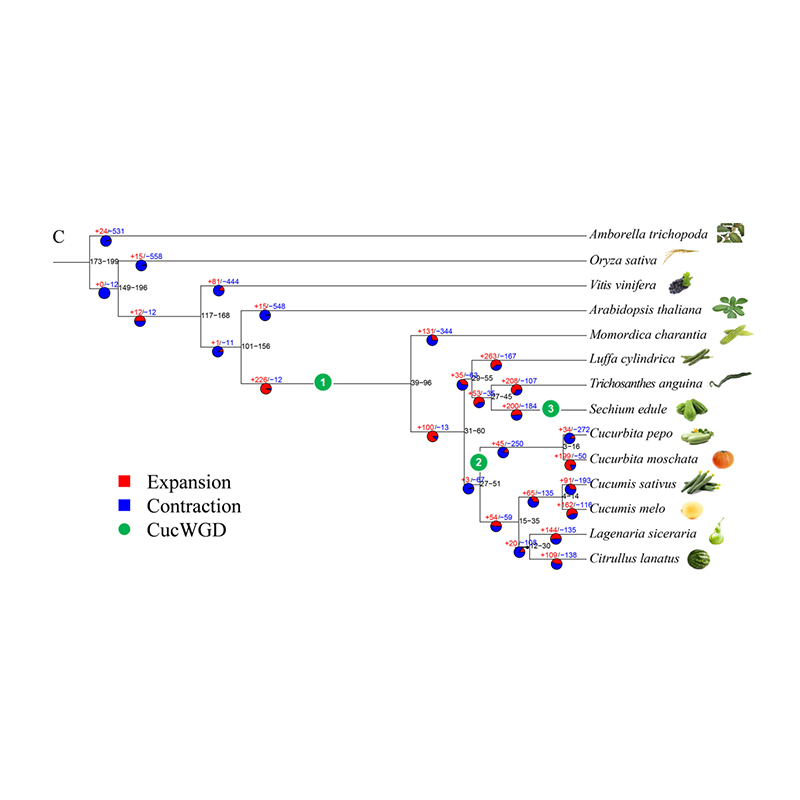
2. Fylogeni och genfamiljeanalys på chayote (Sechium edule): Genom att analysera chayote och de andra 13 besläktade arterna i genfamiljen visade sig Chayote vara närmast besläktad med ormkalebass (Trichosanthes anguina). Chayote härrörande från ormkalebass i omkring 27-45 Mya och helgenomduplicering (WGD) observerades i chayote i 25±4 Mya, vilket är den tredje WGD-händelsen i cucuibitaceae.
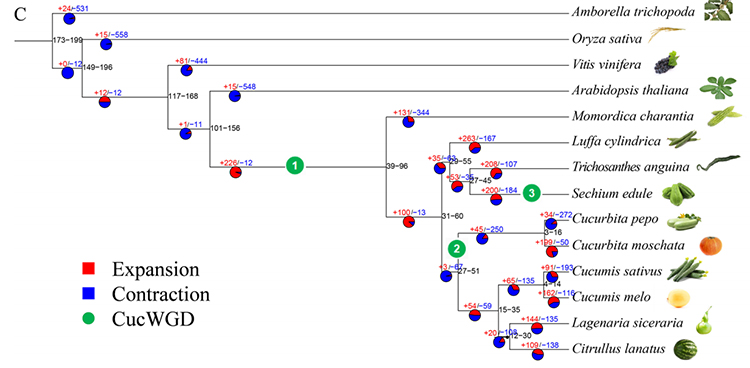
Fu A et al.,Trädgårdsbruksforskning, 2021
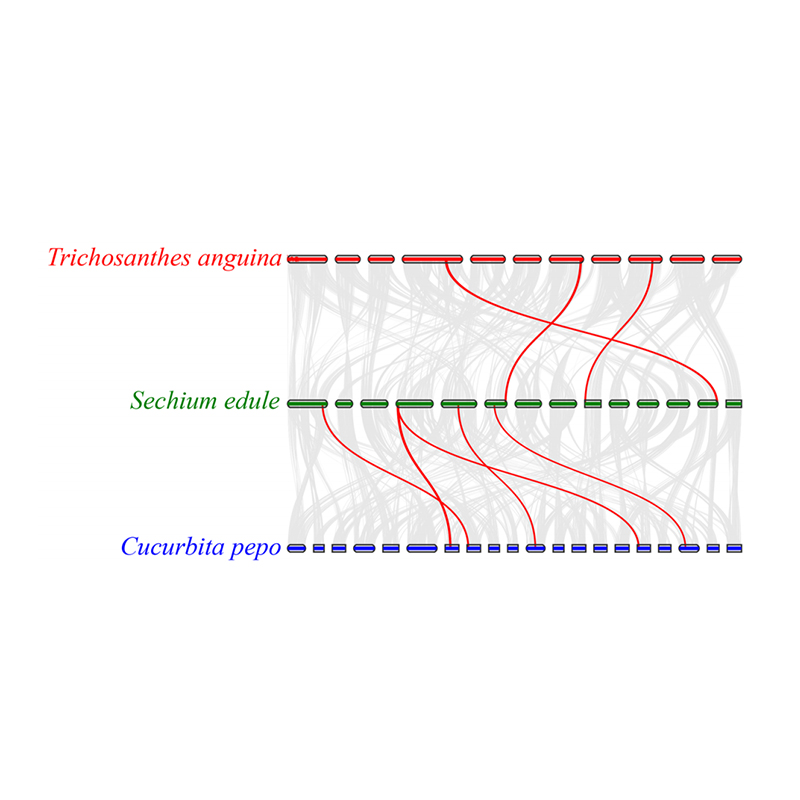
3. Syntenyanalys: Vissa gener relaterade till fytohormoner i fruktutveckling hittades i chayote, ormkalebass och squash. Korrelationen mellan chayote och squash är något högre än den mellan chayote och ormkalebass.
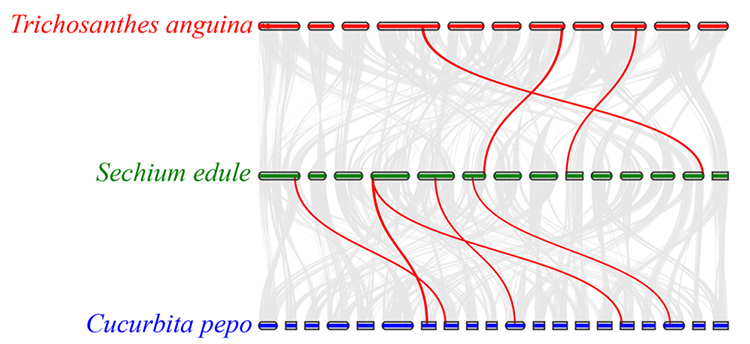
Fu A et al.,Trädgårdsbruksforskning, 2021
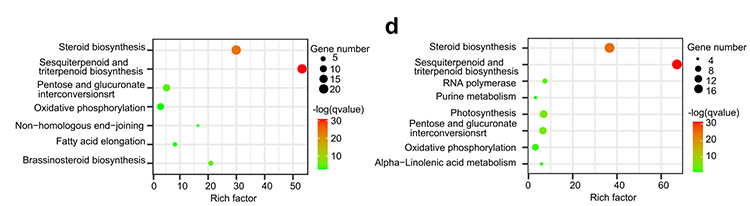
4.Genfamiljanalys: KEGG-berikning på genfamiljens expansion och sammandragning i G.thurberi- och G.davidsonii-genom visade att steroidbiosyntes och brassinosteroidbiosyntesrelaterade gener utökades.
Yang Z et al.,BMC Biologi, 2021
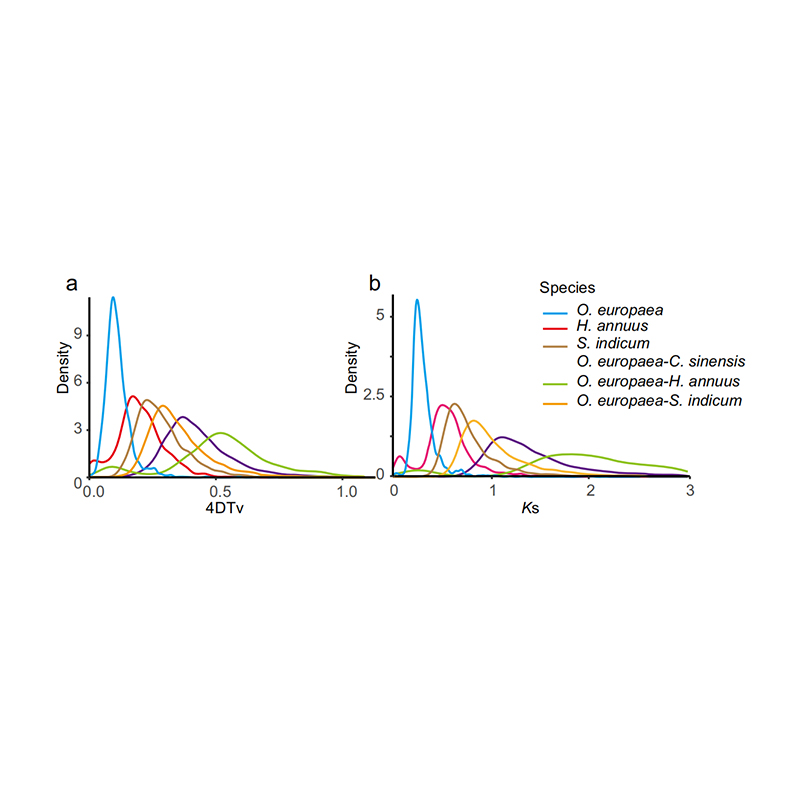
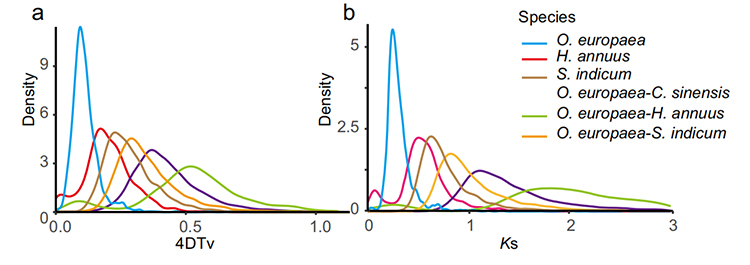
5. Helgenomdupliceringsanalys: 4DTV- och Ks-distributionsanalys visade helgenomdupliceringshändelse. Toppar av intraspecies visade dupliceringshändelser. Toppar mellan arter visade artbildningshändelser. Analysen visade att vid jämförelse med de andra tre närbesläktade arterna gick O. europaea igenom en storskalig genduplicering på senare tid.
Rao G et al.,Trädgårdsbruksforskning, 2021
BMK Fall
Rose utan prickle: genomiska insikter kopplade till fuktanpassning
Publicerad: National Science Review, 2021
Sekvenseringsstrategi:
'BasyesTörnlös' (R.Wichurainan) genom:
Ca. 93 X PacBio + ca. 90 X Nanopore + 267 X Illumina
Nyckelresultat
1. R.wichuraiana-genomet av hög kvalitet konstruerades med hjälp av långlästa sekvenseringstekniker, vilket ger en sammansättning på 530,07 Mb (uppskattad genomstorlek var cirka 525,9 Mb genom flödescytometri och 525,5 genom genomundersökning; Heterozygositeten var cirka 1,03%). BUSCOs uppskattade poäng var 93,9 %. Jämfört med "Old blush" (haploOB), bekräftades kvaliteten och fullständigheten av detta genom av basenbasnoggrannhet och LTR-sammansättningsindex (LAI=20,03). R.wichuriana-genomet innehåller 32 674 proteinkodande gener.
2.Multi-omics gemensam analys, bestående av jämförande genomik, transkriptomik, QTL-analys av genetisk population, avslöjade den avgörande artbildningen mellan R. wichuraiana och Rosa chinensis. Expressionsvariation av relaterade gener i QTL var också sannolikt associerad med stamprickle-mönster.
Jämförande genomikanalys mellan Basye;s Thornless och Rosa chinensis inklusive syntenyanalys, genfamiljekluster, expansions- och kontraktionsanalys, avslöjade ett stort antal variationer, som relaterade till avgörande egenskaper hos rosor. Den unika expansionen i NAC- och FAR1/FRS-genfamiljen var mycket sannolikt associerad med resistens mot svarta fläckar.
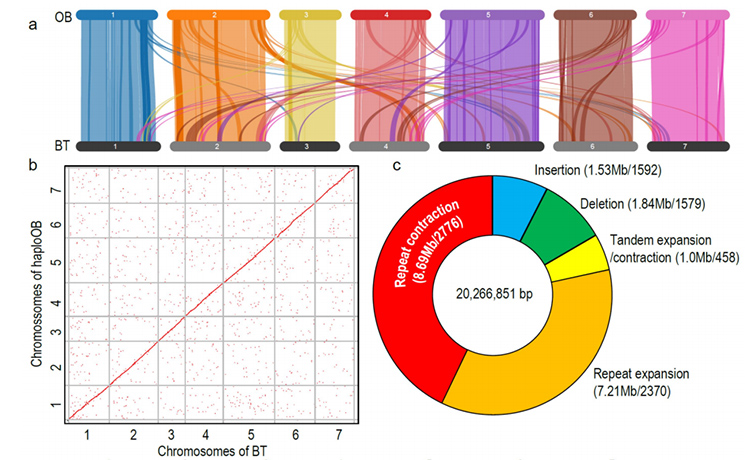

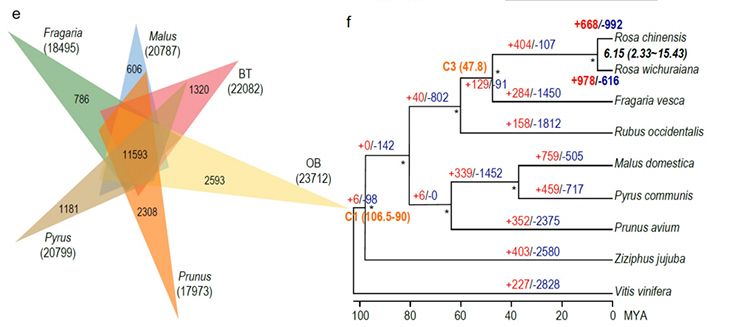
Jämförande genomikanalys mellan BT- och haploOB-genom.
Zhong, M., et al. "Ros utan prickle: genomiska insikter kopplade till fuktanpassning"National Science Review, 2021;, nwab092.





