

Transcriptoma Espacial BMKMANU S1000

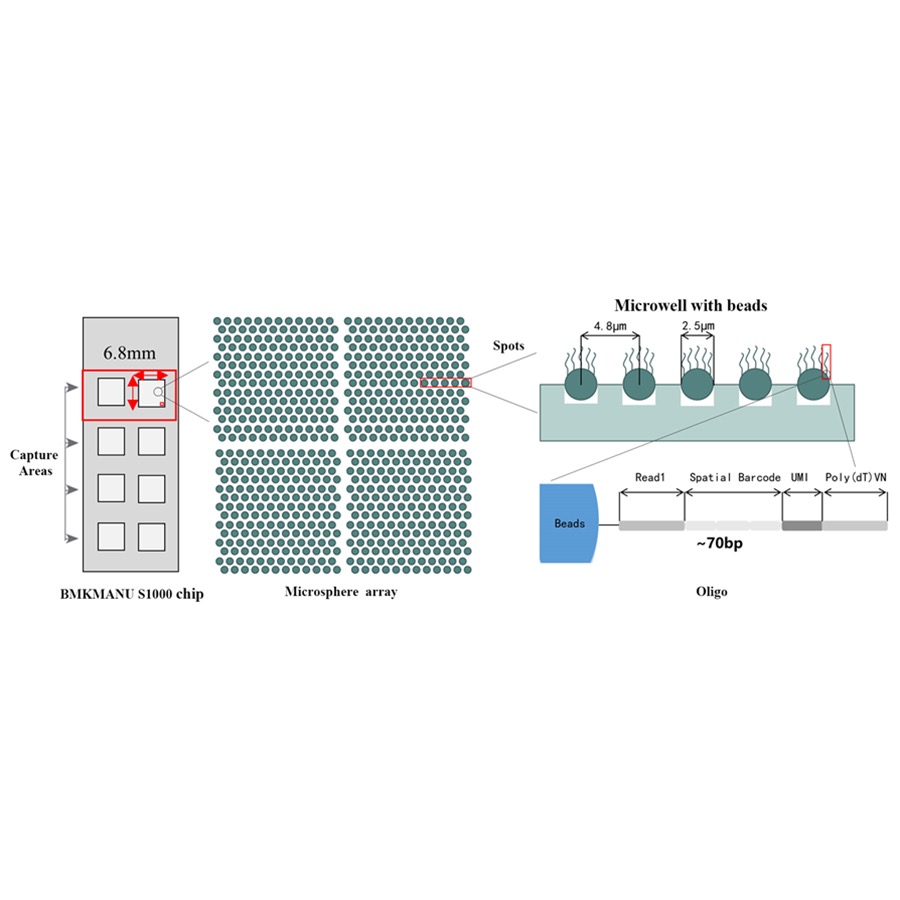
Esquema Técnico do Transcriptoma Espacial BMKMANU S1000

Características
● Resolução: 5 µM
● Diâmetro do ponto: 2,5 µM
● Número de vagas: aproximadamente 2 milhões
● 3 formatos de área de captura possíveis: 6,8 mm * 6,8 mm, 11 mm * 11 mm ou 15 mm * 20 mm
● Cada esfera com código de barras é carregada com primers compostos por 4 seções:
cauda poli (dT) para iniciação de mRNA e síntese de cDNA
Identificador Molecular Único (UMI) para corrigir o viés de amplificação
Código de barras espacial
Sequência de ligação do primer de sequenciamento de leitura parcial 1
● H&E e coloração fluorescente de cortes
● Possibilidade de usotecnologia de segmentação celular: integração de coloração H&E, coloração fluorescente e sequenciamento de RNA para determinar os limites de cada célula e atribuir corretamente a expressão gênica a cada célula.
Vantagens do BMKMANU S1000
●Resolução subcelular: Cada área de captura continha >2 milhões de pontos espaciais com código de barras com diâmetro de 2,5 µm e espaçamento de 5 µm entre centros de pontos, permitindo a análise do transcriptoma espacial com resolução subcelular (5 µm).

●Análise de resolução multinível:Análise flexível em vários níveis variando de 100 μm a 5 μm para resolver diversas características do tecido com resolução ideal.

●Possibilidade de usar tecnologia de segmentação celular “Três em um slide”:Combinando coloração de fluorescência, coloração H&E e sequenciamento de RNA em uma única lâmina, nosso algoritmo de análise "três em um" permite a identificação de limites celulares para transcriptômica subsequente baseada em células.

●Compatível com múltiplas plataformas de sequenciamento: Estão disponíveis sequenciamento NGS e de leitura longa.
●Design flexível de 1 a 8 áreas de captura ativa: O tamanho da área de captura é flexível, sendo possível utilizar 3 formatos (6,8 mm * 6,8 mm., 11 mm * 11 mm e 15 mm * 20 mm)
●Serviço completo: Ele integra todas as etapas baseadas em experiência e habilidade, incluindo crio-secção, coloração, otimização de tecidos, código de barras espacial, preparação de biblioteca, sequenciamento e bioinformática.
●Bioinformática abrangente e visualização de resultados fácil de usar:o pacote inclui 29 análises e mais de 100 números de alta qualidade, combinados com o uso de software desenvolvido internamente para visualizar e personalizar a divisão de células e agrupamento pontual.
●Análise e visualização de dados personalizada: disponível para diferentes solicitações de pesquisa
●Equipe Técnica Altamente Qualificada: com experiência em mais de 250 tipos de tecidos e mais de 100 espécies, incluindo humanos, camundongos, mamíferos, peixes e plantas.
●Atualizações em tempo real sobre todo o projeto: com controle total do progresso experimental.
●Análise Conjunta Opcional com Sequenciamento de mRNA de célula única
Especificações de serviço
|
Amostra Requisitos
| Biblioteca |
Estratégia de sequenciamento
| Dados recomendados | Controle de qualidade |
| Amostras criogênicas incorporadas em OCT, 3 blocos por amostra | Biblioteca de cDNA S1000 | Illumina PE150 (outras plataformas disponíveis) | 100 mil leituras PE por 100 uM (60-150GB) | RIS>7 |
Para obter mais detalhes sobre orientações de preparação de amostras e fluxo de trabalho de serviço, sinta-se à vontade para falar com umEspecialista BMKGENE
Fluxo de trabalho de serviço
Na fase de preparação da amostra, é realizado um teste inicial de extração de RNA em massa para garantir que um RNA de alta qualidade possa ser obtido. Na fase de otimização tecidual, as seções são coradas e visualizadas e as condições de permeabilização para liberação de mRNA do tecido são otimizadas. O protocolo otimizado é então aplicado durante a construção da biblioteca, seguido de sequenciamento e análise de dados.
O fluxo de trabalho completo do serviço envolve atualizações em tempo real e confirmações do cliente para manter um ciclo de feedback responsivo, garantindo uma execução tranquila do projeto.


Os dados gerados pelo BMKMANU S1000 são analisados por meio do software “BSTMatrix”, desenvolvido de forma independente pela BMKGENE, gerando uma Matriz de Expressão Gênica. A partir daí, é gerado um relatório padrão que inclui controle de qualidade de dados, análise de amostra interna e análise intergrupo.
● Controle de qualidade de dados:
Produção de dados e distribuição do índice de qualidade
Detecção de genes por local
Cobertura de tecido
● Análise de amostra interna:
Riqueza genética
Clustering spot, incluindo análise de dimensão reduzida
Análise de expressão diferencial entre clusters: identificação de genes marcadores
Anotação funcional e enriquecimento de genes marcadores
● Análise intergrupo:
Recombinação de manchas de ambas as amostras (por exemplo, doentes e controle) e reagrupamento
Identificação de genes marcadores para cada cluster
Anotação funcional e enriquecimento de genes marcadores
Expressão Diferencial do mesmo cluster entre grupos
Além disso, o “BSTViewer” desenvolvido pela BMKGENE é uma ferramenta fácil de usar que permite ao usuário visualizar a expressão genética e agrupar pontos em diferentes resoluções.
BMKGene desenvolveu software para visualização amigável
Clustering spot BSTViewer em resolução multinível

BSTCellViewer: divisão de células automática e manual

Análise de amostra interna
Agrupamento pontual:

Identificação de genes marcadores e distribuição espacial:

Análise Intergrupo
Combinação de dados de ambos os grupos e reagrupamento:

Genes marcadores de novos clusters:

Explore os avanços facilitados pelos serviços de transcriptômica espacial da BMKGene com a tecnologia BMKManu S1000 nesta publicação em destaque:
Canção, X. et al. (2023) 'A transcriptômica espacial revela células de clorênquima induzidas pela luz envolvidas na promoção da regeneração de brotos em calos de tomate',Anais da Academia Nacional de Ciências dos Estados Unidos da América, 120(38), pág. e2310163120. doi: 10.1073/pnas.2310163120
Você, Y. et al. (2023) 'Comparação sistemática de métodos transcriptômicos espaciais baseados em sequenciamento',bioRxiv, pág. 2023.12.03.569744. doi: 10.1101/2023.12.03.569744.






{kind=link}