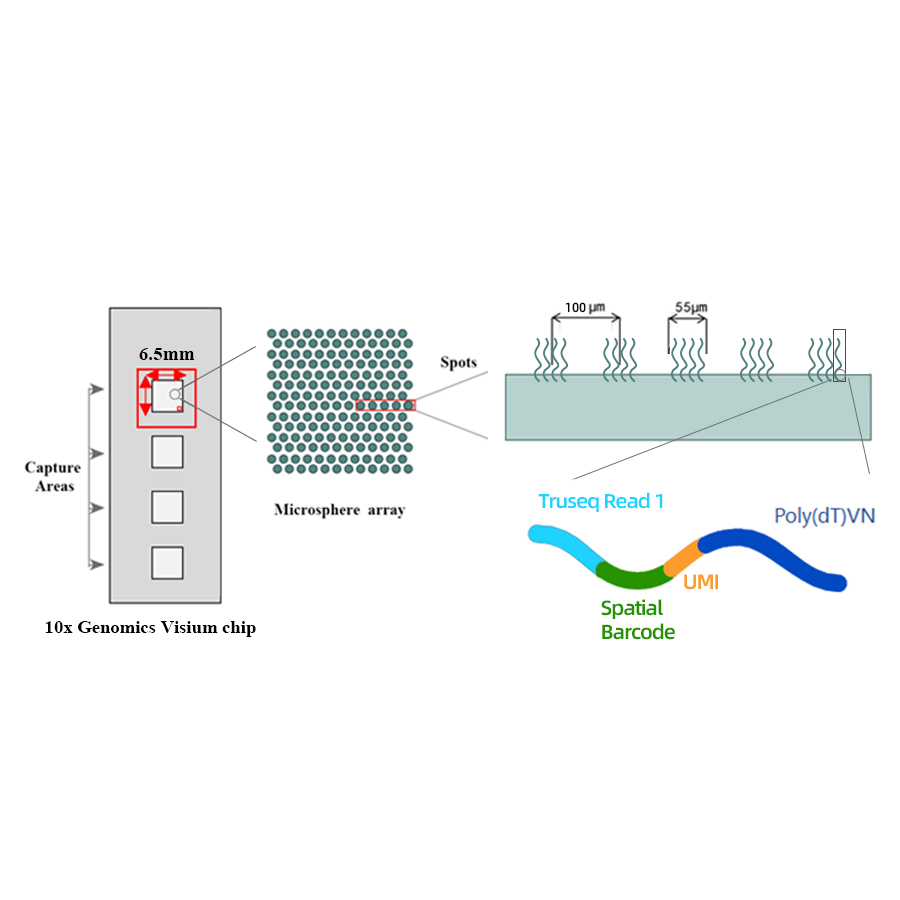
10x Genomics Visium Spatial Transcriptoma

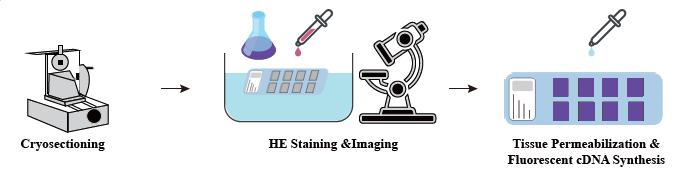
Esquema técnico

Características
● Resolução: 100 µm
● Diâmetro pontual: 55 µm
● Número de pontos: 4992
● Área de captura: 6,5 x 6,5 mm
● Cada ponto com código de barras é carregado com iniciadores compostos por 4 seções:
- Cauda de poli (dt) para priming de mRNA e síntese de cDNA
- Identificador molecular exclusivo (UMI) para corrigir o viés de amplificação
- código de barras espacial
- Sequência de ligação de leitura parcial 1 Primer de seqüenciamento
● coloração de H&E das seções
Vantagens
●Serviço único: integra todas as etapas baseadas em experiência e habilidades, incluindo crio-seção, coloração, otimização de tecidos, código de barras espaciais, preparação da biblioteca, sequenciamento e bioinformática.
● Equipe técnica altamente qualificada: Com experiência em mais de 250 tipos de tecidos e mais de 100 espécies, incluindo humanos, camundongos, mamíferos, peixes e plantas.
●Atualização em tempo real em todo o projeto: com controle total do progresso experimental.
●Bioinformática padrão abrangente:O pacote inclui 29 análises e mais de 100 números de alta qualidade.
●Análise e visualização de dados personalizados: Disponível para diferentes solicitações de pesquisa.
●Análise de articulação opcional com sequenciamento de mRNA de célula única
Especificações
| Requisitos de amostra | Biblioteca | Estratégia de sequenciamento | Dados recomendados | Controle de qualidade |
| Amostras criogênicas incorporadas a outubro (Diâmetro ideal: aprox. 6x6x6 mm³) 2 blocos por amostra | Biblioteca de cDNA de 10x Visium | Illumina PE150 | 50k PE Reads por spot (60 GB) | Rin> 7 |
Para mais detalhes sobre a orientação de preparação de amostras e fluxo de trabalho de serviço, sinta -se à vontade para conversar com umEspecialista em BMKGENE
Fluxo de trabalho de serviço
Na fase de preparação da amostra, um estudo inicial de extração de RNA a granel é realizado para garantir que um RNA de alta qualidade possa ser obtido. No estágio de otimização de tecidos, as seções são manchadas e visualizadas e as condições de permeabilização para liberação de mRNA do tecido são otimizadas. O protocolo otimizado é então aplicado durante a construção da biblioteca, seguido de sequenciamento e análise de dados.
O fluxo de trabalho de serviço completo envolve atualizações em tempo real e confirmações do cliente para manter um loop de feedback responsivo, garantindo a execução suave do projeto.


Inclui a seguinte análise:
Controle de qualidade de dados:
o Saída de dados e distribuição de pontuação de qualidade
o Detecção de genes por ponto
o Cobertura de tecido
Análise de amostra interna:
o riqueza de genes
o Cluster Spot, incluindo análise de dimensão reduzida
o Análise de expressão diferencial entre clusters: identificação de genes marcadores
o Anotação funcional e enriquecimento de genes marcadores
Análise entre grupos
o Re-combinação de manchas de ambas as amostras (por exemplo, doente e controle) e recruta novamente
o Identificação de genes marcadores para cada cluster
o Anotação funcional e enriquecimento de genes marcadores
o Expressão diferencial do mesmo aglomerado entre os grupos
Análise de amostra interna
Cluster spot
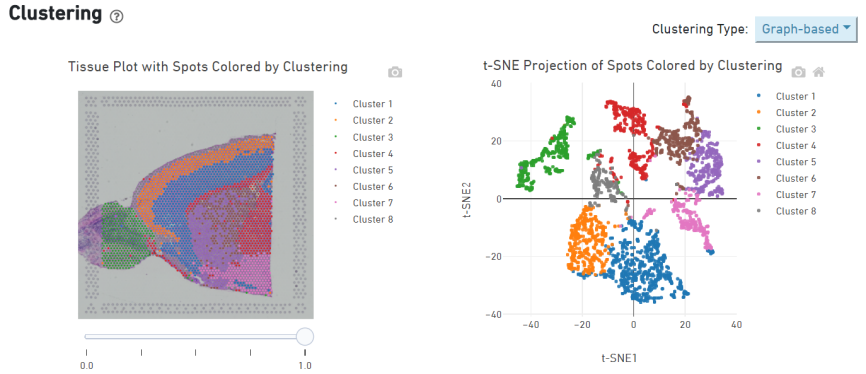
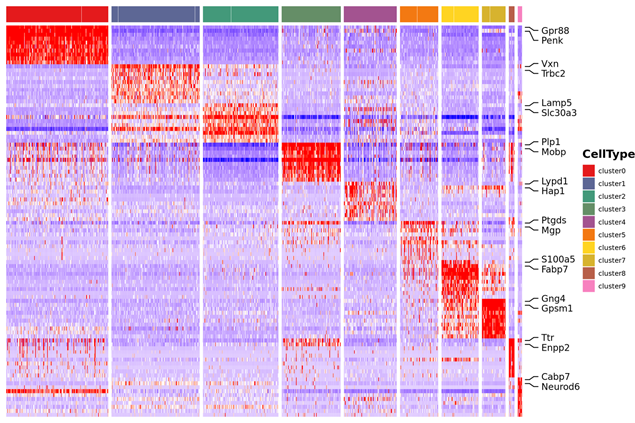
Genes marcadores Identificação e distribuição espacial

Análise entre grupos
Combinação de dados de ambos os grupos e recruta de recruta

Genes marcadores de novos clusters

Explore os avanços facilitados pelo serviço transcriptômico espacial da BMKGENE por 10x Visium nessas publicações em destaque:
Chen, D. et al. (2023) 'MTHL1, um potencial homólogo de Drosophila de GPCRs de adesão de mamíferos, está envolvido em reações antitumorais a células oncogênicas injetadas em moscas',Anais da Academia Nacional de Ciências dos Estados Unidos da América, 120 (30), p. E2303462120. doi: /10.1073/pnas.2303462120
Chen, Y. et al. (2023) 'Aço permite o delineamento de alta resolução de dados transcriptômicos espaço-temporais',Briefings em bioinformática, 24 (2), pp. 1-10. doi: 10.1093/bib/bbad068.
Liu, C. et al. (2022) 'Um atlas espaço -temporal da organogênese no desenvolvimento de flores da orquídea',Pesquisa de ácidos nucleicos, 50 (17), pp. 9724-9737. doi: 10.1093/nar/gkac773.
Wang, J. et al. (2023) 'Integrando a transcriptômica espacial e o seqüenciamento de RNA único-nucleus revela as possíveis estratégias terapêuticas para o leiomioma uterino',Jornal Internacional de Ciências Biológicas, 19 (8), pp. 2515–2530. doi: 10.7150/ijbs.83510.