

Sekwencjonowanie fragmentów wzmacnianych przez specyficzne (SLAF-Seq)

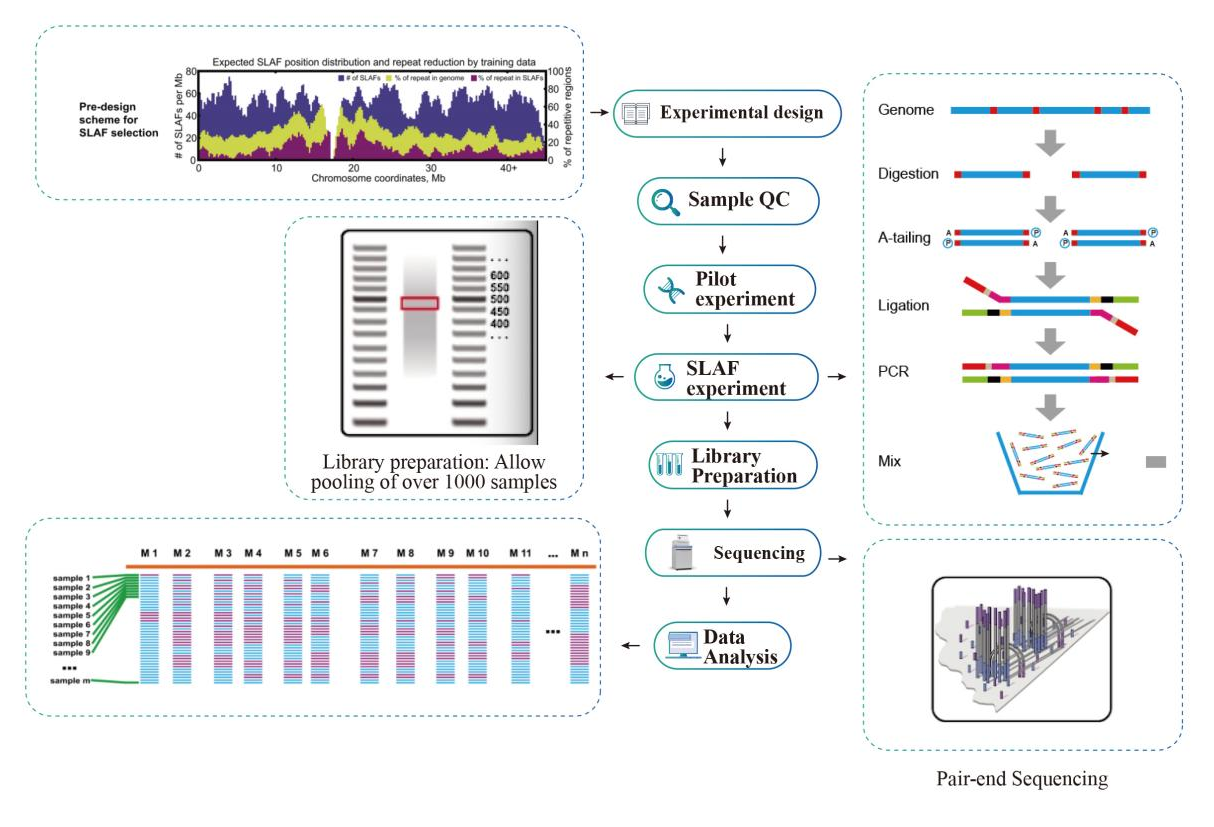
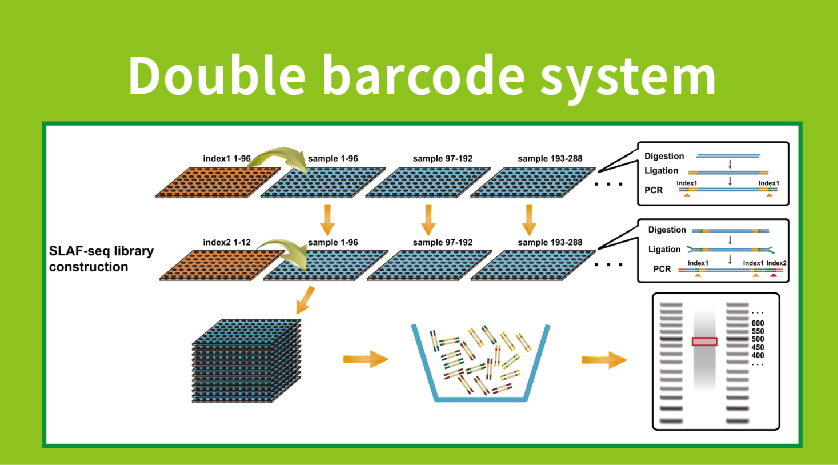
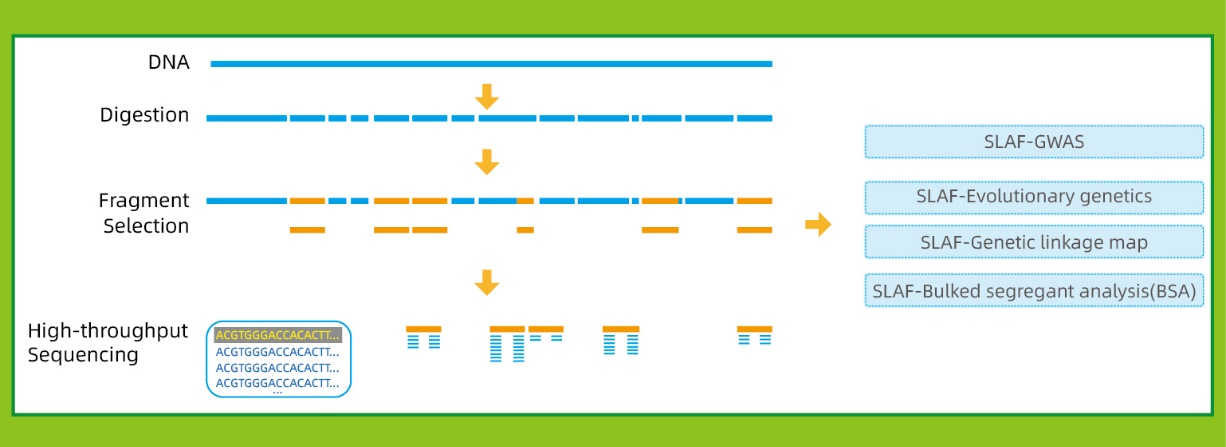
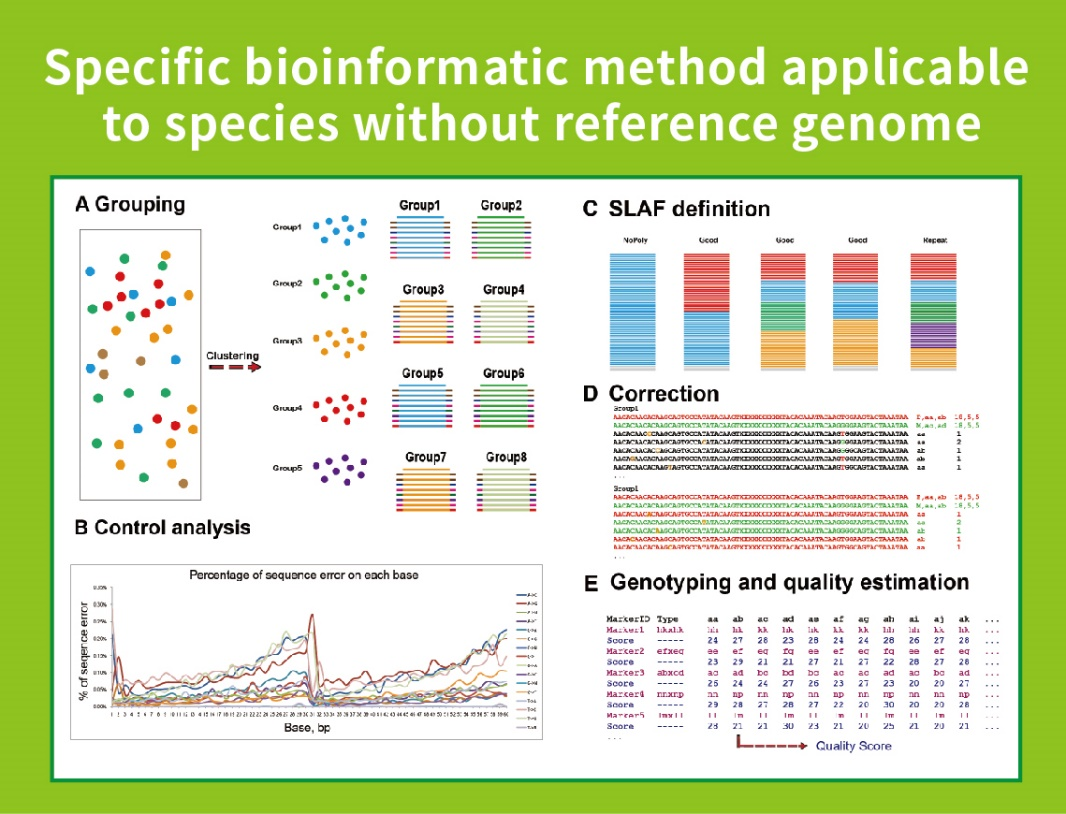
Przepływ pracy

Schemat techniczny

Funkcje serwisowe
● Sekwencjonowanie Novaseq z PE150.
● Przygotowanie biblioteki z podwójnym kodem kreskowym, umożliwiając pulę ponad 1000 próbek.
● Ta technika może być stosowana z genomem referencyjnym lub bez, z różnymi bioinformatycznymi rurociągami dla każdego przypadku:
Z referencyjnym genomem: odkrycie SNP i Indel
Bez genomu referencyjnego: klastrowanie próbki i odkrywanie SNP
● Ww silicoKombinacje enzymów wielokrotnych enzymów wielokrotnych jest badane, aby znaleźć te, które generują jednolity rozkład znaczników SLAF wzdłuż genomu.
● Podczas wstępnego eksperymentu trzy kombinacje enzymów są testowane w 3 próbkach w celu wygenerowania 9 bibliotek SLAF, a informacje te są wykorzystywane do wybrania optymalnej kombinacji enzymu ogranicznika dla projektu.
Zalety serwisowe
●Wysokie odkrycie markerów genetycznych: Integracja wysokoprzepustowego systemu podwójnego kodu kreskowego pozwala na jednoczesne sekwencjonowanie dużych populacji, a wzmocnienie specyficzne dla locus zwiększa wydajność, zapewniając, że liczby tagów spełniają różnorodne wymagania różnych pytań badawczych.
● Niska zależność od genomu: Można go zastosować do gatunków z genomem referencyjnym lub bez.
●Elastyczny projekt schematu: Jedno-enzym, podwójny enzym, trawienie wielu enzymów i różne rodzaje enzymów można wybrać, aby zaspokoić różne cele badawcze lub gatunki. .w silicoPrenerowanie jest przeprowadzane w celu zapewnienia optymalnego projektu enzymu.
● Wysoka wydajność trawienia enzymatycznego: Przewodnictwow silicoWstępne opracowanie i przed eksperymentem zapewniły optymalną konstrukcję z równomiernym rozkładem znaczników SLAF na chromosomie (1 znacznik SLAF/4KB) i zmniejszone sekwencję powtarzającą się (<5%).
●Rozległa wiedza specjalistyczna: Nasz zespół wprowadza bogate doświadczenie w każdym projekcie, z osiągnięciem zamykania ponad 5000 projektów sekcji SLAF na setkach gatunków, w tym roślin, ssakach, ptakach, owadach i organizmach wodnych.
● Samozwańczy bioinformatyczny przepływ pracy: BMKGENE opracował zintegrowany przepływ pracy bioinformatyczny dla SLAF-Seq, aby zapewnić niezawodność i dokładność ostatecznego wyjścia.
Specyfikacje usług
| Rodzaj analizy | Zalecana skala populacji | Strategia sekwencjonowania | |
| Głębokość sekwencjonowania znacznika | Numer znacznika | ||
| Mapy genetyczne | 2 rodziców i> 150 potomstwa | Rodzice: 20x WGS Offsping: 10x | Rozmiar genomu: <400 MB: zalecane jest WGS <1 GB: 100 tys. Tagów 1-2GB :: 200k tagów > 2 GB: 300 tys. Tagów MAX 500K TAGI |
| Badania asocjacyjne całego genomu (GWAS) | ≥200 próbek | 10x | |
| Ewolucja genetyczna | ≥30 próbek, z> 10 próbkami z każdej podgrupy | 10x | |
Wymagania serwisowe
Stężenie ≥ 5 ng/µl
Całkowita ilość ≥ 80 ng
Nanodrop OD260/280 = 1,6-2.5
Żel agarozowy: brak lub ograniczona degradacja lub zanieczyszczenie
Zalecana dostawa próbki
Pojemnik: 2 ml rurki wirówki
(W przypadku większości próbek zalecamy nie zachowanie w etanolu)
Etykietowanie próbki: Próbki muszą być wyraźnie oznaczone i identyczne z przesłanym formularzem informacji przykładowych.
Wysyłka: suche lody: Próbki należy najpierw pakować w torby i pochować w suchym lodzie.
Przepływ pracy serwisowej






Próbka QC
Eksperyment pilotażowy
Eksperyment SLAF
Przygotowanie biblioteki
Sekwencjonowanie
Analiza danych
Usługi po sprzedaży
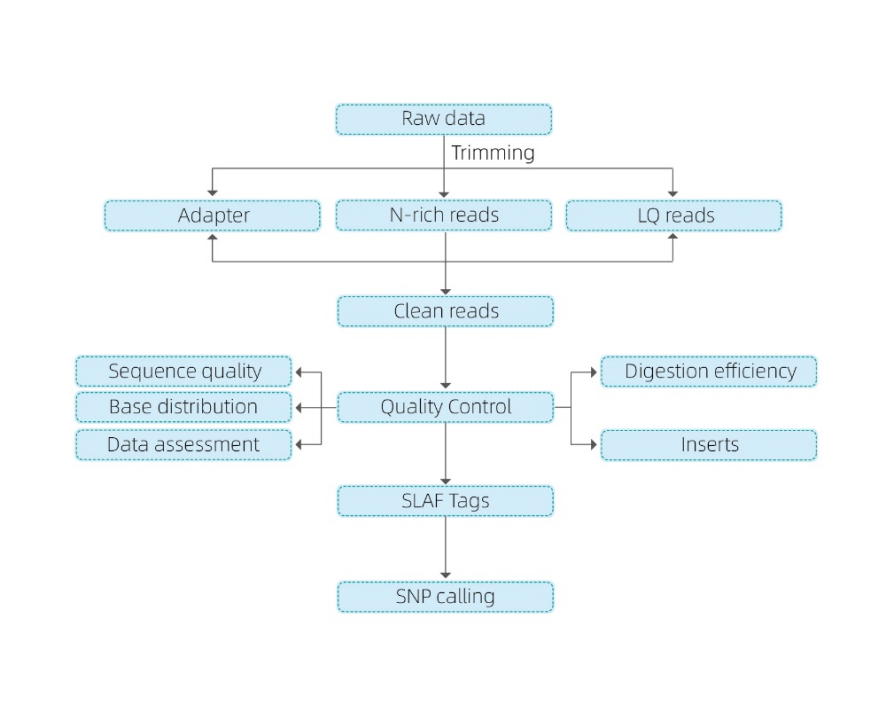 Obejmuje następującą analizę:
Obejmuje następującą analizę:

- Sekwencjonowanie danych QC
- Rozwój tagów SLAF
Mapowanie na genom referencyjny
Bez genomu referencyjnego: grupowanie
- Analiza tagów SLAF.: Statystyka, dystrybucja w całym genomie
- Odkrycie markerów: SNP, Indel, SNV, CV Calling and Adnotation
Rozkład znaczników SLAF na chromosomach:

Rozkład SNP na chromosomach:
 Adnotacja SNP
Adnotacja SNP

| Rok | Dziennik | IF | Tytuł | Zastosowania |
| 2022 | Komunikacja natury | 17.694 | Genomowe podstawy giga-chromosomów i genomu giga drzewnego Paeonia Ostii | SLAF-GWAS |
| 2015 | Nowy fitolog | 7.433 | Ślady udomowienia Kotwiczne regiony genomowe o znaczeniu agronomicznym w Soja | SLAF-GWAS |
| 2022 | Journal of Advanced Research | 12.822 | Sztuczne introgresje gossypium w całym genomie Gossypium Barbadense do G. hirsutum Ujawnij doskonałe loci w celu jednoczesnej poprawy jakości i plonu włókien bawełnianych cechy | Genetyka ewolucyjna SLAF |
| 2019 | Roślina molekularna | 10.81 | Analiza genomowa populacji i montaż de novo ujawniają pochodzenie chwastu Ryż jako gra ewolucyjna | Genetyka ewolucyjna SLAF |
| 2019 | Nature Genetics | 31.616 | Sekwencja genomu i różnorodność genetyczna wspólnego karpia, Cyprinus carpio | Mapa SLAF-Linkage |
| 2014 | Nature Genetics | 25.455 | Genom uprawianych orzeszków ziemnych zapewnia wgląd w kariotypy roślin strączkowych, poliploid Ewolucja i udomowienie upraw. | Mapa SLAF-Linkage |
| 2022 | Plant Biotechnology Journal | 9.803 | Identyfikacja ST1 ujawnia selekcję obejmującą autostopem morfologii nasion i zawartość ropy podczas udomowienia soi | Rozwój rynku SLAF |
| 2022 | International Journal of Molecular Sciences | 6.208 | Identyfikacja i rozwój markera DNA dla Mollis Mollis 2NS (2d) Disomic Chromosom Subition | Rozwój rynku SLAF |
| Rok | Dziennik | IF | Tytuł | Zastosowania |
| 2023 | Granice w nauce roślinnej | 6.735 | Mapowanie QTL i analiza transkryptomu zawartości cukru podczas dojrzewania owoców pirus piryfolia | Mapa genetyczna |
| 2022 | Plant Biotechnology Journal | 8.154 | Identyfikacja ST1 ujawnia wybór obejmujący autostop morfologii nasion i zawartość oleju podczas udomowienia soi
| Wzywa SNP |
| 2022 | Granice w nauce roślinnej | 6.623 | Mapowanie skojarzeń w całym genomie ledwo fenotypy w środowisku suszy.
| GWas |





