

Sekwencjonowanie genomu roślin/zwierząt de novo





De Nowosekwencjonowanie odnosi się do konstrukcji całego genomu gatunku przy użyciu technologii sekwencjonowania w przypadku braku genomu referencyjnego. Wprowadzenie i powszechne przyjęcie sekwencjonowania trzeciej generacji, obejmującego dłuższe odczyty, znacznie usprawniło składanie genomu poprzez zwiększenie nakładania się odczytów. To ulepszenie jest szczególnie istotne w przypadku trudnych genomów, takich jak te wykazujące wysoką heterozygotyczność, wysoki stosunek regionów powtarzalnych, poliploidalnych i regionów z elementami powtarzalnymi, nieprawidłową zawartość GC lub dużą złożoność, które są zazwyczaj słabo złożone przy użyciu sekwencjonowania z krótkim odczytem sam.
Nasze kompleksowe rozwiązanie zapewnia zintegrowane usługi sekwencjonowania i analizy bioinformatyczne, które zapewniają wysokiej jakości złożony genom de novo. Wstępne badanie genomu za pomocą Illuminy pozwala oszacować wielkość i złożoność genomu, a informacje te służą do poprowadzenia kolejnego etapu sekwencjonowania z długim odczytem za pomocą PacBio HiFi, a następnieod nowamontaż kontigów. Późniejsze zastosowanie składania HiC umożliwia zakotwiczenie kontigów w genomie, uzyskując montaż na poziomie chromosomu. Wreszcie genom jest opisywany poprzez przewidywanie genów i sekwencjonowanie genów ulegających ekspresji, odwołując się do transkryptomów z krótkim i długim odczytem.
Funkcje usługi
● Integracja wielu usług sekwencjonowania i bioinformatycznych w jednym rozwiązaniu:
Badanie genomu za pomocą firmy Illumina w celu oszacowania rozmiaru genomu i wybrania dalszych kroków;
Długo czytana sekwencja dlaod nowamontaż kontigów;
Sekwencjonowanie Hi-C w celu zakotwiczenia chromosomu;
sekwencjonowanie mRNA w celu adnotacji genów;
Walidacja montażu.
● Usługa odpowiednia do konstruowania nowych genomów lub ulepszania istniejących genomów referencyjnych dla interesujących gatunków.
Zalety serwisu
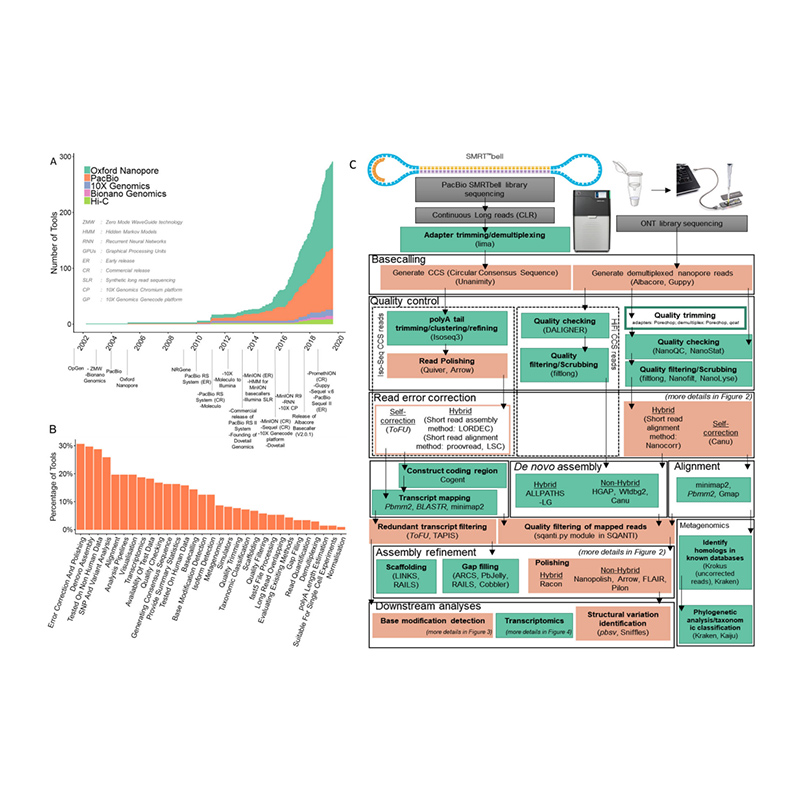
Rozwój platform sekwencjonowania i bioinformatyki wod nowaskładanie genomu
(Amarasinghe SL i in.,Biologia genomu, 2020)
●Obszerna wiedza specjalistyczna i dorobek publikacyjny: BMKGene zgromadziło ogromne doświadczenie w składaniu wysokiej jakości genomów różnych gatunków, w tym genomów diploidalnych i bardzo złożonych genomów gatunków poliploidalnych i allopoliploidalnych. Od 2018 roku przyczyniliśmy się do ponad300 publikacji o dużym wpływie, z czego ponad 20 zostało opublikowanych w Nature Genetics.
● Kompleksowe rozwiązanie: nasze zintegrowane podejście łączy wiele technologii sekwencjonowania i analiz bioinformatycznych w spójny przepływ pracy, zapewniając wysokiej jakości złożony genom.
●Dopasowane do Twoich potrzeb: Przepływ pracy w ramach naszych usług można dostosować, co pozwala na adaptację genomów o różnorodnych cechach i specyficznych potrzebach badawczych. Obejmuje to przystosowanie się do genomów gigantycznych, genomów poliploidalnych, genomów wysoce heterozygotycznych i nie tylko.
●Wysoko wykwalifikowany zespół bioinformatyczny i laboratoryjny: z dużym doświadczeniem zarówno w obszarze eksperymentalnym, jak i bioinformatycznym w zakresie złożonych zespołów genomu oraz szeregiem patentów i praw autorskich do oprogramowania.
●Wsparcie posprzedażowe:Nasze zaangażowanie wykracza poza realizację projektu i obejmuje 3-miesięczny okres obsługi posprzedażnej. W tym czasie oferujemy monitorowanie projektu, pomoc w rozwiązywaniu problemów oraz sesje pytań i odpowiedzi, aby odpowiedzieć na wszelkie pytania związane z wynikami.
Specyfikacje usług
| Badanie genomu | Składanie genomu | Poziom chromosomów | Adnotacja genomu |
| 50X Illumina NovaSeq PE150
| 30X odczytów PacBio CCS HiFi | 100X Hi-C | Sekwencja RNA Illumina PE150 10 Gb + (fakultatywny) Pełnej długości sekwencja RNA PacBio 40 Gb lub Nanopor 12 Gb |
Wymagania serwisowe
W przypadku badania genomu, składania genomu i składania Hi-C:
| Tkankowe lub ekstrahowane kwasy nukleinowe | Badanie genomu | Składanie genomu za pomocą PacBio | Zespół Hi-C |
| Wnętrzności zwierzęce | 0,5-1 g
| ≥ 3,5 g | ≥2 g |
| Mięsień zwierzęcy | ≥ 5 g | ||
| Krew ssaków | 1,5 ml
| ≥ 5 ml | ≥2 ml |
| Krew drobiowa/rybna | ≥ 0,5 ml | ||
| Roślina - Świeży Liść | 1-2 gr | ≥ 5 g | ≥ 4 g |
| Hodowane komórki |
| ≥ 1x108 | ≥ 1x107 |
| Owad | 0,5-1 g | ≥ 3 g | ≥ 2 g |
| Wyekstrahowane DNA | Stężenie: ≥1 ng/µl Ilość ≥ 30 ng Ograniczona lub żadna degradacja lub zanieczyszczenie | Stężenie: ≥ 50 ng/µl Ilość: 10 µg/komórkę przepływową/próbkę OD260/280=1,7-2,2 OD260/230=1,8-2,5 Ograniczona lub żadna degradacja lub zanieczyszczenie |
-
|
W przypadku adnotacji genomu za pomocą transkryptomiki:
| Tkankowe lub ekstrahowane kwasy nukleinowe | Transkryptom Illuminy | Transkryptom PacBio | Transkryptom nanoporów |
| Roślina – korzeń/łodyga/płatek | 450 mg | 600 mg | |
| Roślina – Liść/Nasiono | 300 mg | 300 mg | |
| Roślina - Owoc | 1,2 g | 1,2 g | |
| Serce/jelito zwierzęcia | 300 mg | 300 mg | |
| Wnętrzności/mózg zwierząt | 240 mg | 240 mg | |
| Mięsień zwierzęcy | 450 mg | 450 mg | |
| Kości zwierzęce/włosy/skóra | 1 gr | 1 gr | |
| Stawonogi - Owad | 6 | 6 | |
| Stawonogi -Skorupia | 300 mg | 300 mg | |
| Pełna krew | 1 tuba | 1 tuba | |
| Wyekstrahowany RNA | Stężenie: ≥ 20 ng/µl Ilość ≥ 0,3 µg OD260/280=1,7-2,5 OD260/230=0,5-2,5 RIN≥ 6 5≥28S/18S≥1 | Stężenie: ≥ 100 ng/µl Ilość ≥ 0,75 µg OD260/280=1,7-2,5 OD260/230=0,5-2,5 RIN≥ 8 5≥28S/18S≥1 | Stężenie: ≥ 100 ng/µl Ilość ≥ 0,75 µg OD260/280=1,7-2,5 OD260/230=0,5-2,5 RIN≥ 7,5 5≥28S/18S≥1 |
Zalecana dostawa próbek
Pojemnik: probówka wirówkowa o pojemności 2 ml (nie zaleca się stosowania folii aluminiowej)
(W przypadku większości próbek zalecamy nie konserwować ich w etanolu.)
Etykietowanie próbek: Próbki muszą być wyraźnie oznakowane i identyczne z przesłanym formularzem informacyjnym dotyczącym próbek.
Wysyłka: Suchy lód: Próbki należy najpierw zapakować w worki i zakopać w suchym lodzie.
Przepływ pracy

Przebieg prac serwisowych

Projekt eksperymentu

Dostawa próbek

Ekstrakcja DNA

Budowa biblioteki

Sekwencjonowanie

Analiza danych

Usługi posprzedażowe

Pełna analiza bioinformatyczna podzielona na 4 etapy:
1) Badanie genomu, oparte na analizie k-merów za pomocą NGS, brzmi:
Ocena wielkości genomu
Ocena heterozygotyczności
Estymacja regionów powtarzalnych
2) Składanie genomu za pomocą PacBio HiFi:
Od nowamontaż
Ocena złożenia: w tym analiza BUSCO pod kątem kompletności genomu i mapowanie odczytów NGS i PacBio HiFi
3) Montaż Hi-C:
Biblioteka Hi-C QC: ocena ważnych interakcji Hi-C
Montaż Hi-C: grupowanie kontigów w grupy, a następnie porządkowanie kontigów w każdej grupie i przypisywanie orientacji kontigów
Ocena Hi-C
4) Adnotacja genomu:
Przewidywanie niekodującego RNA
Identyfikacja sekwencji powtarzalnych (transpozony i powtórzenia tandemowe)
Przewidywanie genów
§Od nowa: algorytmy ab initio
§ W oparciu o homologię
§ Na podstawie transkryptomu, z długimi i krótkimi odczytami: odczyty sąod nowazmontowane lub zmapowane do projektu genomu
§ Adnotacja przewidywanych genów w wielu bazach danych
1) Badanie genomu – analiza k-merów

2) Składanie genomu

2) Montaż genomu – PacBio HiFi odczytuje mapowanie do projektu zestawu

2) Montaż Hi-C – estymacja ważnych par interakcji Hi-C

3) Ocena Hi-C po montażu

4) Adnotacja genomu – integracja przewidywanych genów

4) Adnotacja genomu – adnotacja przewidywanych genów

Poznaj postępy możliwe dzięki usługom składania genomu de novo BMKGene poprzez wyselekcjonowany zbiór publikacji:
Li, C. i in. (2021) „Sekwencje genomu ujawniają globalne trasy rozprzestrzeniania się i sugerują zbieżne adaptacje genetyczne w ewolucji koników morskich”, Nature Communications, 12(1). doi: 10.1038/S41467-021-21379-X.
Li, Y. i in. (2023) „Zmiany chromosomalne na dużą skalę prowadzą do zmian w ekspresji na poziomie genomu, adaptacji środowiskowej i specjacji u gejów (Bos frontalis)”, Molecular Biology and Evolution, 40(1). doi: 10.1093/MOLBEV/MSAD006.
Tian, T. i in. (2023) „Składanie genomu i rozwarstwienie genetyczne wybitnej, odpornej na suszę plazmy zarodkowej kukurydzy”, Nature Genetics 2023 55:3, 55(3), s. 496–506. doi: 10.1038/s41588-023-01297-y.
Zhang, F. i in. (2023) „Ujawnienie ewolucji biosyntezy alkaloidów tropanowych poprzez analizę dwóch genomów w rodzinie Solanaceae”, Nature Communications 2023 14:1, 14(1), s. 1–18. doi: 10.1038/s41467-023-37133-4.
Trudne studia przypadków:
Zespół telomer-telomer:Fu, A. i in. (2023) „Złożenie genomu gorzkiego melona (Momordica charantia L. var. abbreviata Ser.) od telomerów do telomerów ujawnia cechy genetyczne rozwoju, składu i dojrzewania owoców”, Horticulture Research, 10(1). doi: 10.1093/HR/UHAC228.
Montaż haplotypu:Hu, W. i in. (2021) „Genom zdefiniowany przez allele ujawnia różnicowanie bialleliczne podczas ewolucji manioku”, Molecular Plant, 14(6), s. 851–854. doi: 10.1016/j.molp.2021.04.009.
Gigantyczny montaż genomu:Yuan, J. i in. (2022) „Genomic base of the giga-chromosomy i giga-genome of drzewiastej piwonii Paeonia ostii”, Nature Communications 2022 13:1, 13(1), s. 1–16. doi: 10.1038/s41467-022-35063-1.
Zespół genomu poliploidalnego:Zhang, Q. i in. (2022) „Genomic wgląd w niedawną redukcję chromosomów autopoliploidalnej trzciny cukrowej Saccharum spontaneum”, Nature Genetics 2022 54:6, 54(6), s. 885–896. doi: 10.1038/s41588-022-01084-1.





