

Analiza asocjacji całego genomu

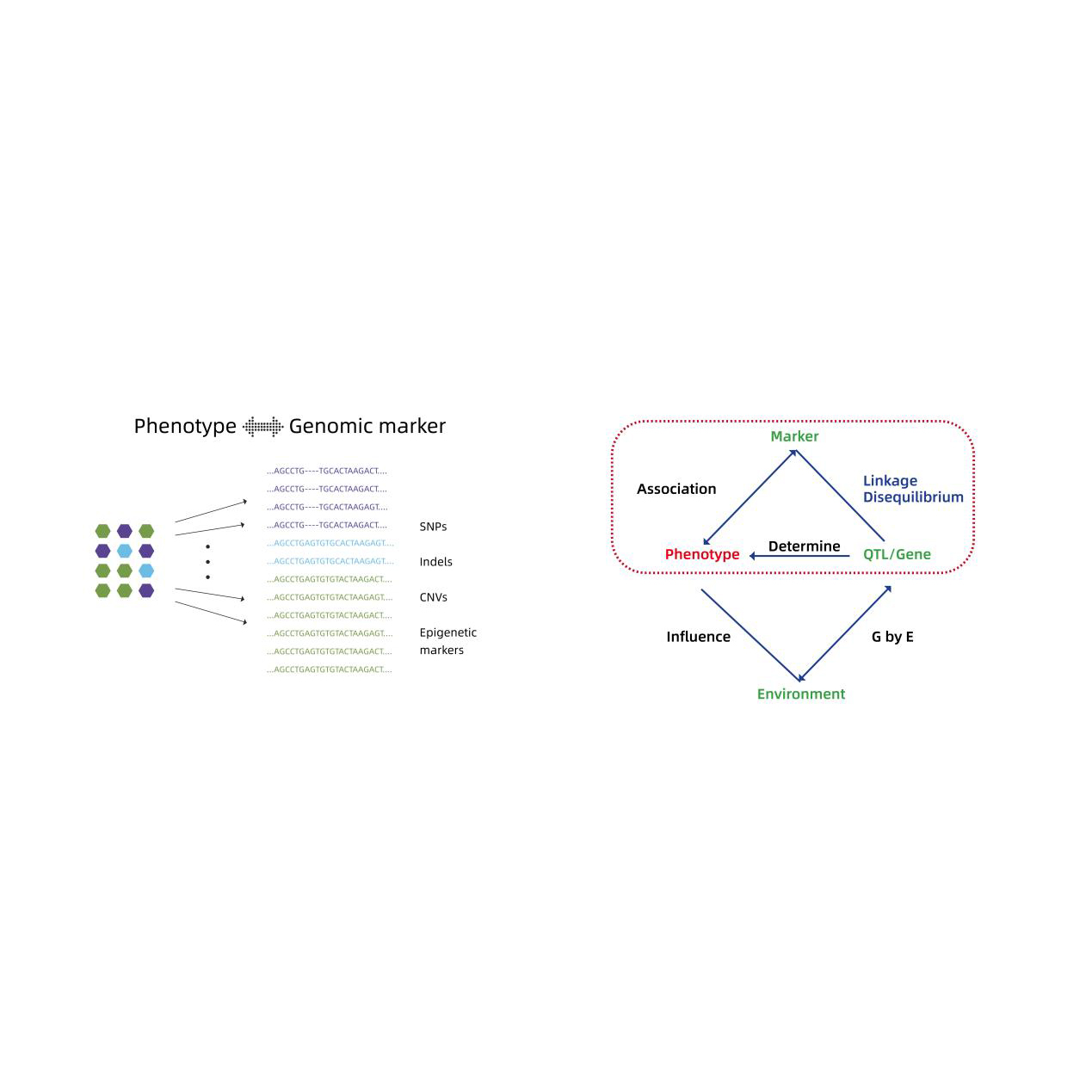
Przepływ pracy

Zalety serwisu
●Obszerna dokumentacja ekspercka i publikacyjna: dzięki zgromadzonemu doświadczeniu w GWAS firma BMKGene zrealizowała setki projektów gatunkowych w badaniach populacyjnych GWAS, pomogła naukowcom w opublikowaniu ponad 100 artykułów, a skumulowany współczynnik wpływu osiągnął 500.
● Kompleksowa analiza bioinformatyczna: przepływ pracy obejmuje analizę powiązań cech SNP, dostarczającą zestaw genów kandydujących i odpowiadającą im adnotację funkcjonalną.
●Wysoko wykwalifikowany zespół bioinformatyków i krótki cykl analityczny: dzięki dużemu doświadczeniu w zaawansowanej analizie genomiki zespół BMKGene zapewnia kompleksowe analizy w krótkim czasie realizacji.
●Wsparcie posprzedażowe:Nasze zaangażowanie wykracza poza realizację projektu i obejmuje 3-miesięczny okres obsługi posprzedażnej. W tym czasie oferujemy monitorowanie projektu, pomoc w rozwiązywaniu problemów oraz sesje pytań i odpowiedzi, aby odpowiedzieć na wszelkie pytania związane z wynikami.
Specyfikacje i wymagania usług
| Rodzaj sekwencjonowania | Zalecana skala populacji | Strategia sekwencjonowania | Wymagania dotyczące nukleotydów |
| Sekwencjonowanie całego genomu | 200 próbek | 10x | Stężenie: ≥ 1 ng/µl Całkowita ilość ≥ 30ng Ograniczona lub żadna degradacja lub zanieczyszczenie |
| Fragment amplifikowany w specyficznym locus (SLAF) | Głębokość znacznika: 10x Liczba tagów: < 400 Mb: zaleca się WGS < 1 Gb: 100 tys. tagów 1Gb > 2 Gb: 300 tys. tagów Maks. 500 tys. tagów | Stężenie ≥ 5 ng/µL Całkowita ilość ≥ 80 ng Nanokropla OD260/280=1,6-2,5 Żel agarozowy: brak lub ograniczona degradacja lub zanieczyszczenie
|
Wybór materiału



Różne odmiany, podgatunki, rasy lokalne/banki genów/rodziny mieszane/dzikie zasoby
Różne odmiany, podgatunki, rasy lądowe
Rodzina półrodzeństwa/rodzina pełnego rodzeństwa/dzikie zasoby
Przebieg prac serwisowych

Projekt eksperymentu

Dostawa próbek

Ekstrakcja RNA

Budowa biblioteki

Sekwencjonowanie

Analiza danych

Usługi posprzedażowe

Zawiera następującą analizę:
- Analiza asocjacji całego genomu: model LM, LMM, EMMAX, FASTLMM
- Adnotacja funkcjonalna genów kandydujących
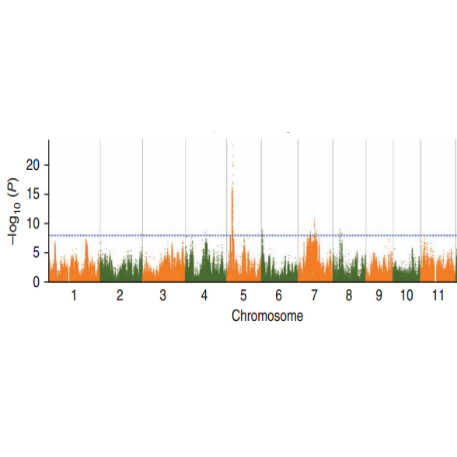
Analiza asocjacji cech SNP – wykres Manhattan

Analiza asocjacji cech SNP – wykres QQ

Poznaj postępy możliwe dzięki usługom de GWAS firmy BMKGene poprzez wyselekcjonowany zbiór publikacji:
Lv, L. i in. (2023) „Wgląd w podstawy genetyczne tolerancji amoniaku u małży brzytwowej Sinonovacula constricta na podstawie badania asocjacyjnego obejmującego cały genom”,Akwakultura, 569, s. 739351. doi: 10.1016/J.AQUACULTURE.2023.739351.
Li, X. i in. (2022) „Analizy multiomiczne 398 posiewów prosa wyczyszczonego ujawniają regiony genomowe powiązane z udomowieniem, cechami metabolitów i działaniem przeciwzapalnym”,Roślina molekularna, 15(8), s. 1367–1383. doi: 10.1016/j.molp.2022.07.003.
Li, J. i in. (2022) „Mapowanie asocjacyjne obejmujące cały genom fenotypów ledwie pozbawionych łusek w środowisku suszy”,Granice w nauce o roślinach, 13, s. 924892. doi: 10.3389/FPLS.2022.924892/BIBTEX.
Zhao, X. i in. (2021) „GmST1, który koduje sulfotransferazę, nadaje oporność na szczepy wirusa mozaiki soi G2 i G3”,Roślina, komórka i środowisko, 44(8), s. 2777–2792. doi: 10.1111/PCE.14066.





