

Genomika porównawcza

Zalety serwisowe
●Obszerna wiedza specjalistyczna i dokumentacja publikacji: Dzięki nagromadzeniu BMKGENE ukończył ponad 90 porównawczych projektów genomicznych, a skumulowany współczynnik uderzenia osiągnął 900.
●Kompleksowa analiza bioinformatyczna: Pakiet analiz zawiera osiem najczęściej wymaganych analiz, zapewniając dobrze zaprojektowane dane gotowe do wydania i pozwalają na łatwą interpretację wyników
●Wysoko wykwalifikowany zespół bioinformatyki i krótki cykl analizy: Przy wielkim doświadczeniu w analizie porównawczej genomiki zespół BMKGENE spełnia różnorodne spersonalizowane wymagania analizy w krótkim czasie
●Wsparcie po sprzedaży:Nasze zobowiązanie wykracza poza ukończenie projektu z 3-miesięcznym okresem usług po sprzedaży. W tym czasie oferujemy obserwację projektu, pomoc w rozwiązywaniu problemów oraz sesje pytań i odpowiedzi, aby rozwiązać wszelkie zapytania związane z wynikami.
Specyfikacje usług
| Szacowany czas skrętu | Liczba gatunków | Ćwiczenie |
| 30 dni roboczych | 6 - 12 | Klastrowanie rodziny genów Ekspansja i skurcz rodziny genów Filogenetyczna konstrukcja drzewa Oszacowanie czasu rozbieżności (wymagana kalibracja kopalna) Czas wstawiania LTR (dla roślin) Duplikacja całego genomu (dla roślin) Ciśnienie selektywne Analiza synteny |
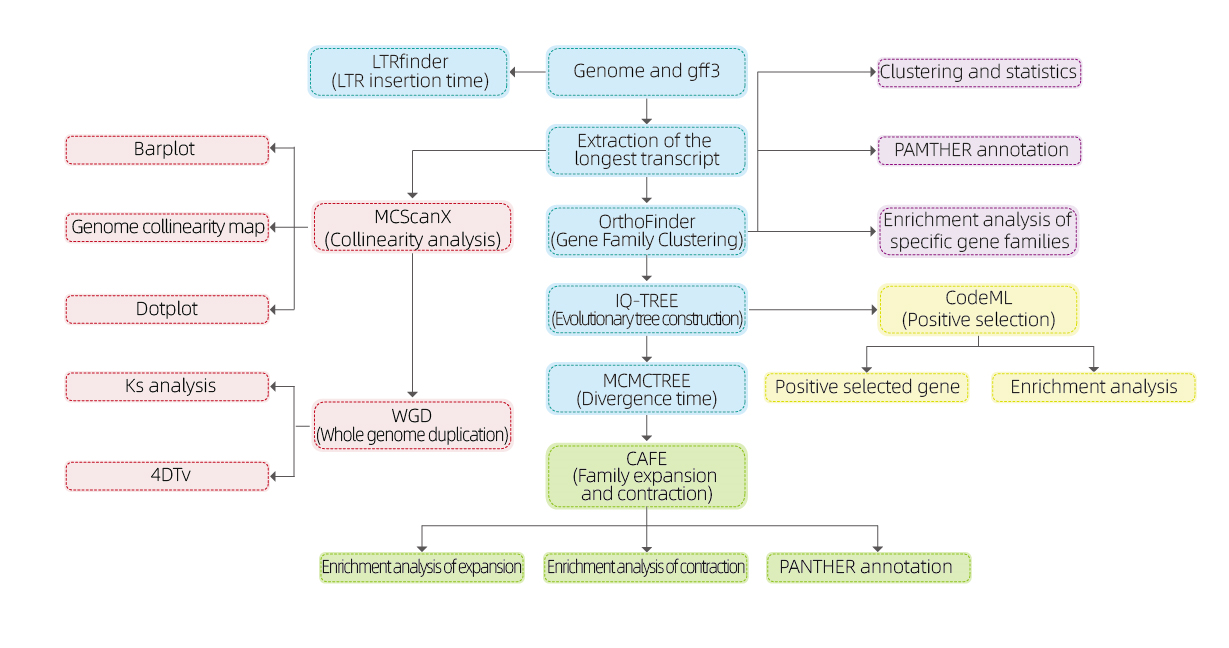
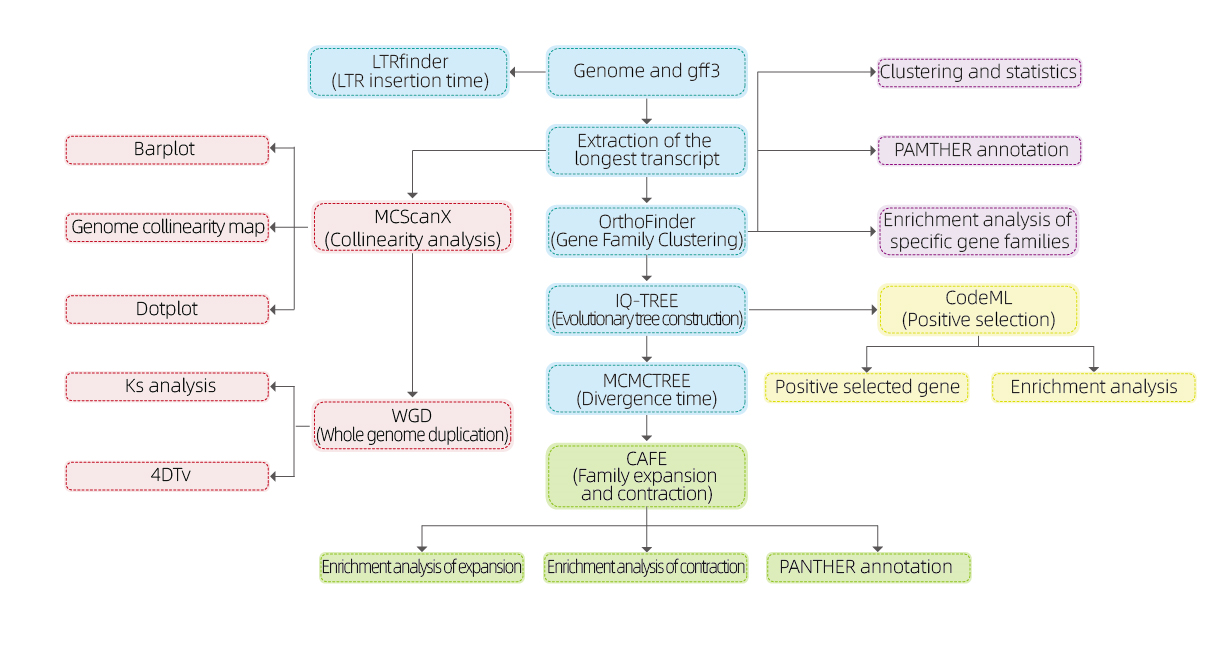
Analizy bioinformatyczne
● Rodzina genów
● Filogenetyka
● Czas rozbieżności
● Presja selektywna
● Analiza synteny
Przykładowe wymagania i dostawa
Przykładowe wymagania:
Tkanka lub DNA do sekwencjonowania i montażu genomu
Dla tkanki
| Gatunek | Tkanka | Ankieta | Pacbio CCS |
| Zwierzę | Tkanka trzewna | 0,5 ~ 1 g | ≥ 3,5 g |
| Tkanka mięśniowa | |||
| ≥ 5,0 g | |||
| ≥ 5,0 ml | |||
| Krew ssaków | |||
| ≥ 0,5 ml | |||
| Krew drobiu/ryb | |||
| Zakład | Świeży liść | 1 ~ 2 g | ≥ 5,0 g |
| Płatek/łodyga | 1 ~ 2 g | ≥ 10,0 g | |
| Korzeń/ziarno | 1 ~ 2 g | ≥ 20,0 g | |
| Komórki | Hodowana komórka | - | ≥ 1 x 108 |
Dane
Pliki sekwencji genomu (.FastA) i pliki adnotacyjne (.GFF3) blisko spokrewnionych gatunków
Przepływ pracy usług

Projekt eksperymentu

Dostawa próbki

Konstrukcja biblioteki

Sekwencjonowanie

Analiza danych

Usługi po sprzedaży
*Wyniki demo pokazane tutaj wszystkie z genomów opublikowanych z Biomarker Technologies
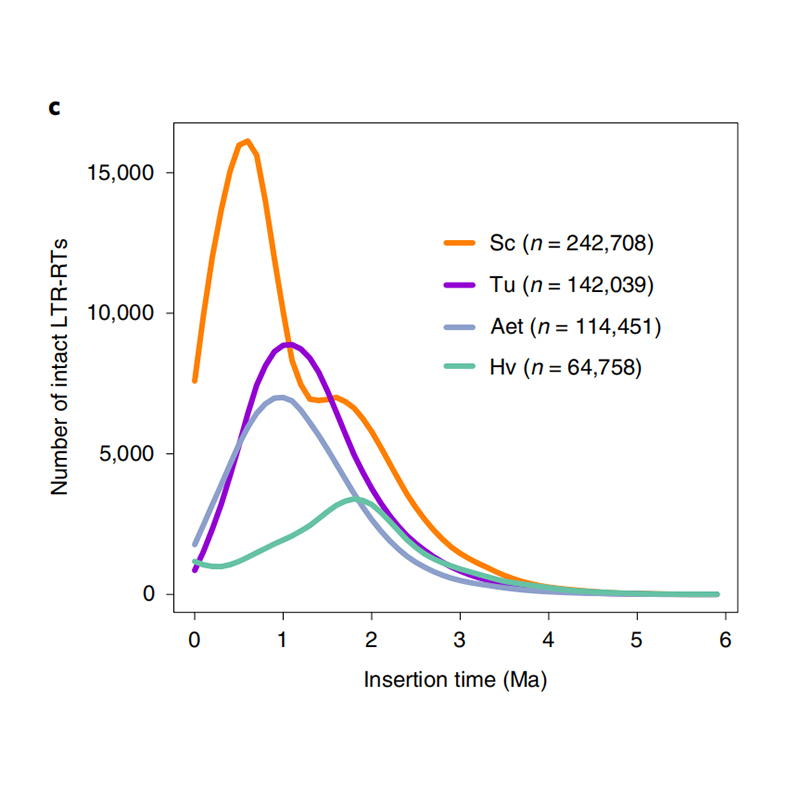
1. Oszacowanie czasu wkładania LTR: Rysunek pokazano unikalny rozkład bimodalny w czasach wstawienia LTR-RTS w genomie Rye, w porównaniu z innymi gatunkami. Ostatni szczyt pojawił się około 0,5 miliona lat temu.
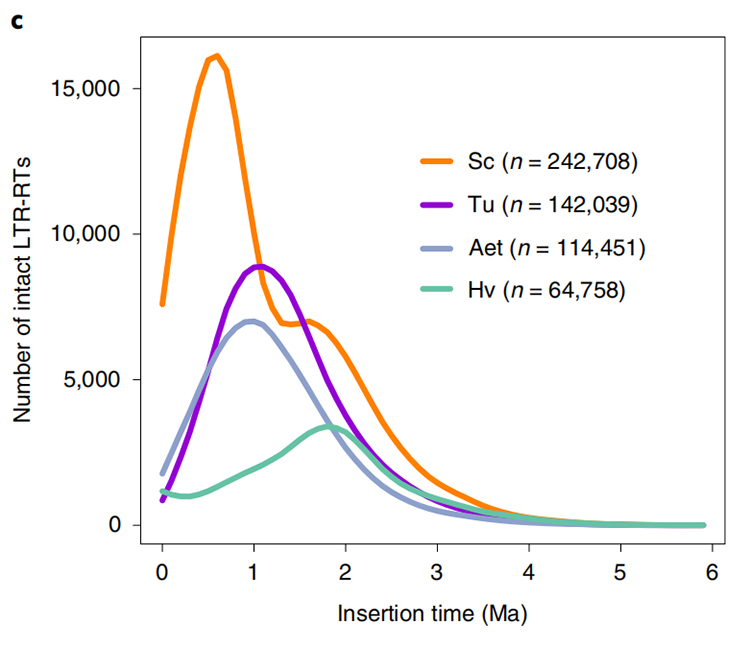
Li Guang i in.,Nature Genetics, 2021
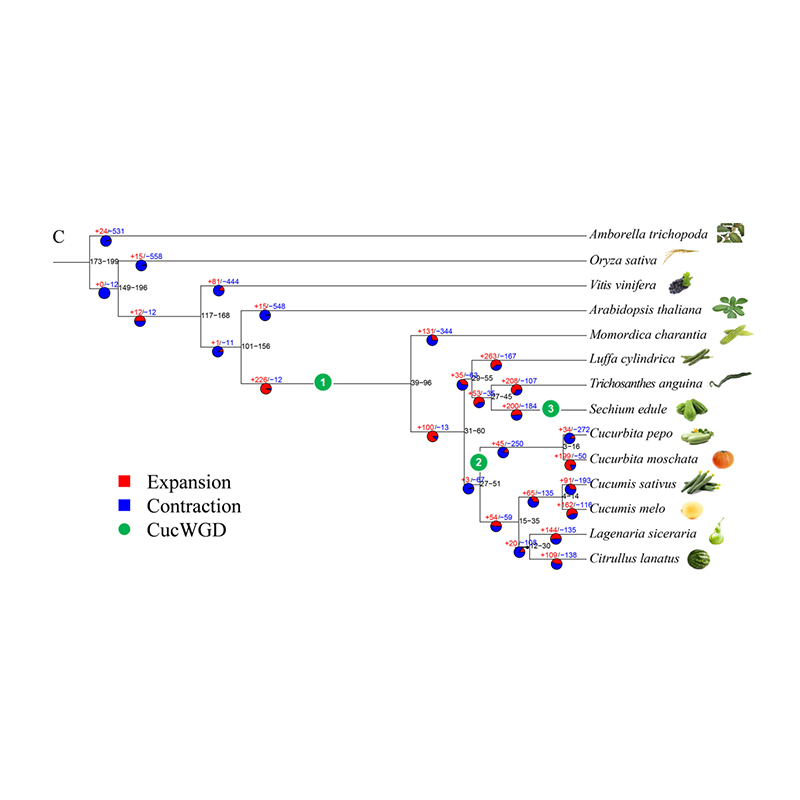
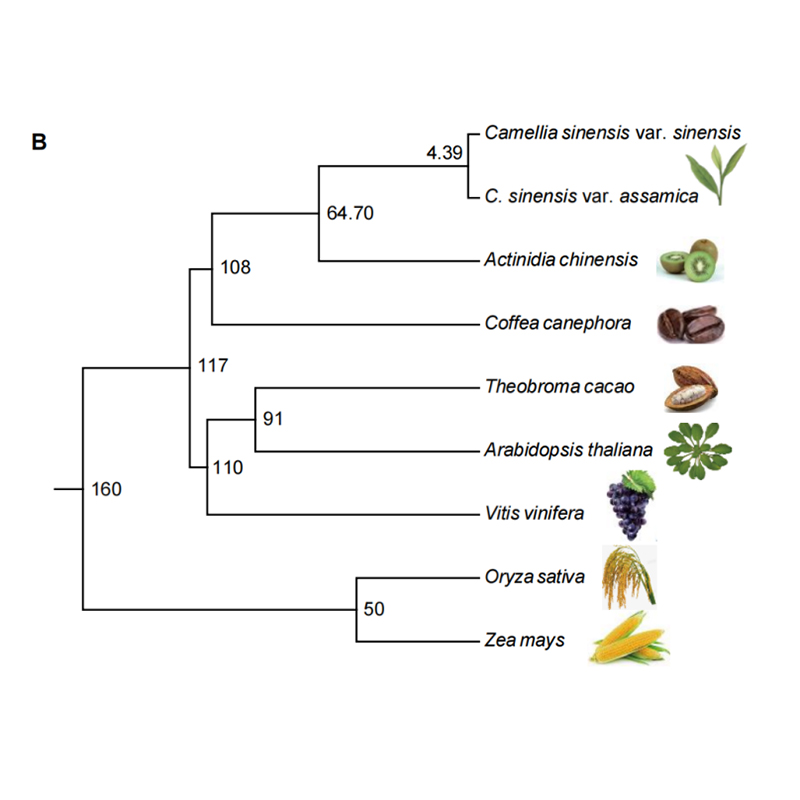
2. Analiza rodziny filogenezji i genów na Chayote (Sechium Edule): Analizując Chayote i pozostałe 13 powiązanych gatunków w rodzinie genów, okazało się, że Chayote był najbardziej powiązany z tykwą Snake (Trichosanthes Anguina). Chayote pochodzący z Gurwy Snake w około 27-45 MyA i powielanie całego genomu (WGD) zaobserwowano w Chayote w 25 ± 4 MyA, który jest trzecim zdarzeniem WGD w Cucuibitaceae.
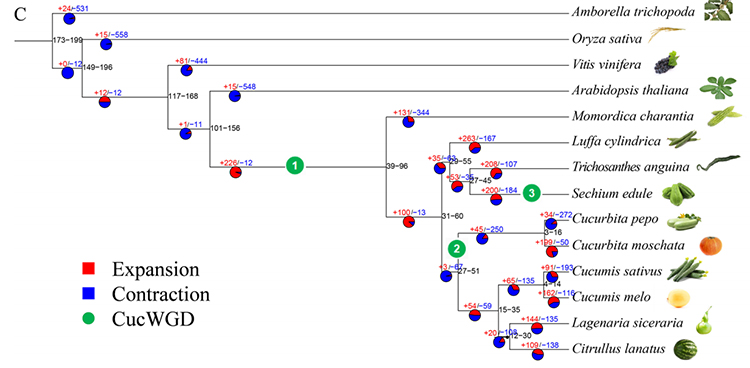
Fu A i in.,Badania ogrodnictwa, 2021
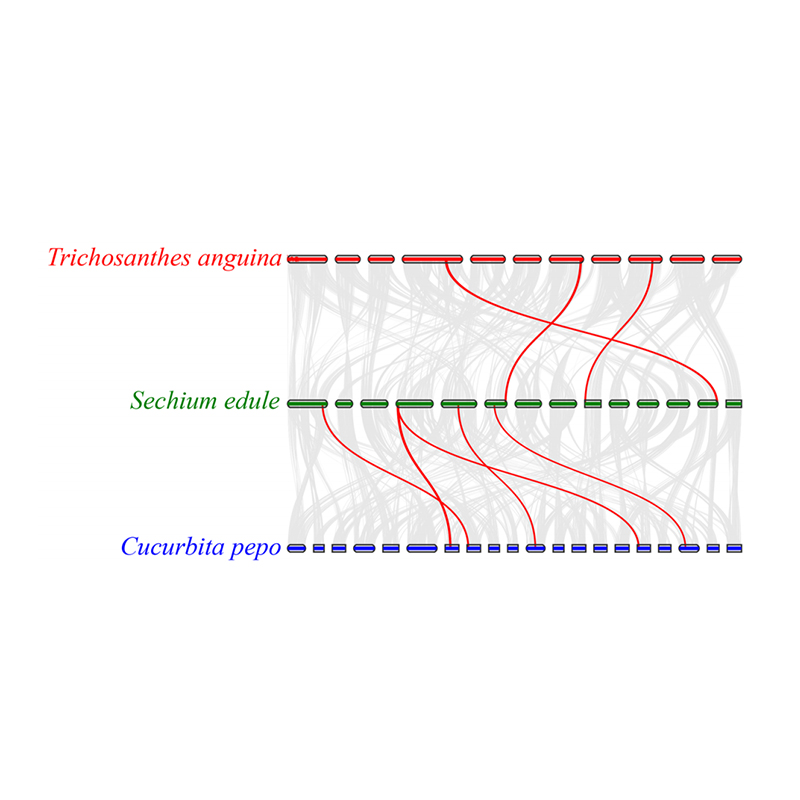
3. Analiza Synteny: Niektóre geny związane z fitohormonami w rozwoju owoców znaleziono w Chayote, Gurd Snake i Squash. Korelacja między chayote a squash jest nieco wyższa niż między Chayote i Gurd Snake.
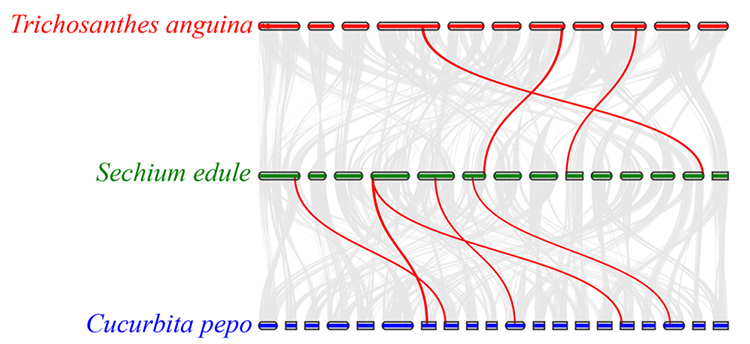
Fu A i in.,Badania ogrodnictwa, 2021
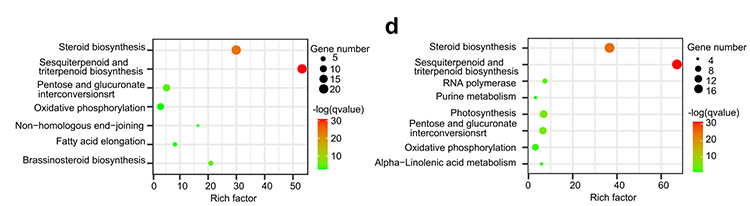
4. Analiza rodziny genetycznej: Wzbogacanie Kegg na ekspansję i skurcz rodziny genów w genomach G.Thurberi i G.Davidsonii wykazały, że geny związane z biosyntezą związanymi z biosyntezą i biosyntezą mosinosteroidów.
Yang Z i in.,BMC Biology, 2021
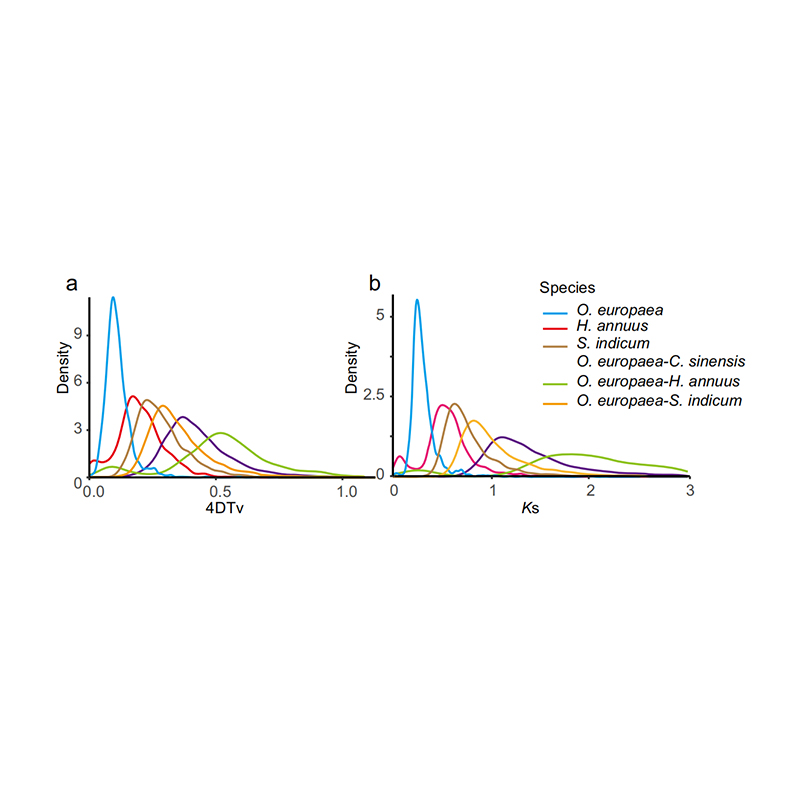
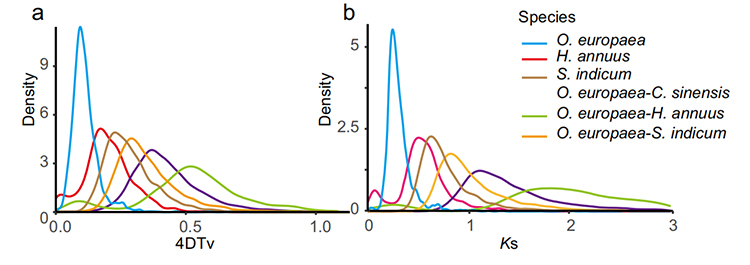
5. Analiza duplikacji genomu: Analiza dystrybucji 4DTV i KS pokazała zdarzenie duplikacji całego genomu. Piki wewnątrzgatunkowe pokazały zdarzenia duplikacji. Piki międzygatunków pokazały zdarzenia specjacyjne. Analiza wykazała, że w porównaniu z pozostałymi trzema ściśle powiązanymi gatunkami O. europaea ostatnio przeszedł na dużą duplikację genów.
Rao G i in.,Badania ogrodnictwa, 2021
Przypadek BMK
Róża bez kutasa: genomowe spostrzeżenia związane z adaptacją wilgoci
Opublikowany: National Science Review, 2021
Strategia sekwencjonowania:
„BaseyeBezcierniowy' (R.Wichurainan) Genom:
Ok. 93 x Pacbio + ok. 90 x nanopore + 267 x Illumina
Kluczowe wyniki
1. Wysoką jakość genom R.Wichuraiana skonstruowano przy użyciu technik sekwencjonowania długoterminowego, które dają montaż 530,07 MB (szacowany wielkość genomu wynosił około 525,9 MB za pomocą cytometrii przepływowej i 525,5 według badania genomu ; Heterozyggotyzator wynosił około 1,03%). Szacowany wynik Busco wyniósł 93,9%. W porównaniu z „starym różem” (HaploOB), jakość i kompletność tego genomu potwierdzono przez podstawową dokładność jednopazową i wskaźnik montażu LTR (LAI = 20,03). Genom R.Wichuraiana zawiera 32 674 geny kodujące białka.
2. Wspólna analiza multimiczna, polegająca na genomice porównawczej, transkryptomiki, analizy QTL populacji genetycznej, ujawniła kluczową specjację między R. wchuraiana i Rosa chinensis. Również zmienność ekspresji pokrewnych genów w QTL prawdopodobnie była związana z wzornictwem kłucia łodygi.
Porównawcza genomika Anaysis między Basye; S ROSA Chinensis, w tym analiza synteny, klaster rodzinny, analiza ekspansji i skurczu, ujawniła dużą liczbę zmian, które związane z kluczowymi cechami w różach. Unikalna ekspansja w rodzinie genów NAC i FAR1/FRS bardzo prawdopodobne jest, że będzie związana z odpornością na czarne miejsce.
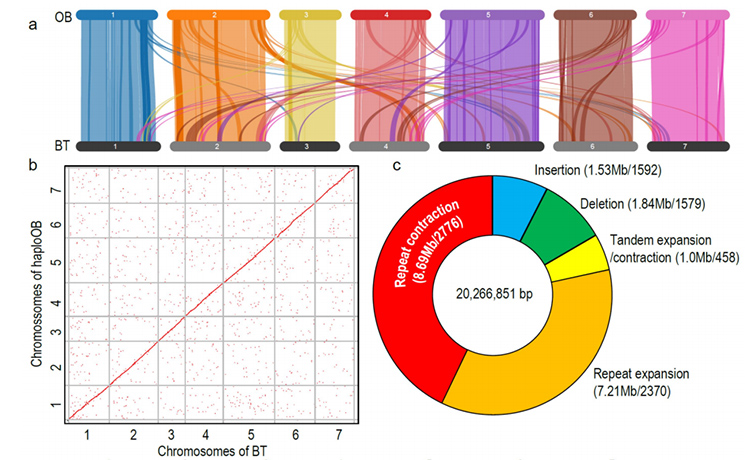

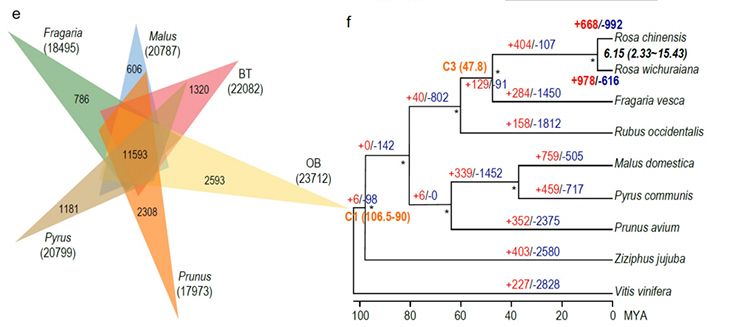
Porównawcza analiza genomiki między genomami BT i HaploOB.
Zhong, M., i in. „Rose bez kutasa: genomowe spostrzeżenia związane z adaptacją wilgoci”National Science Review, 2021;, NWAB092.





