

Analiza zbiorcza segregantów

Zalety serwisowe
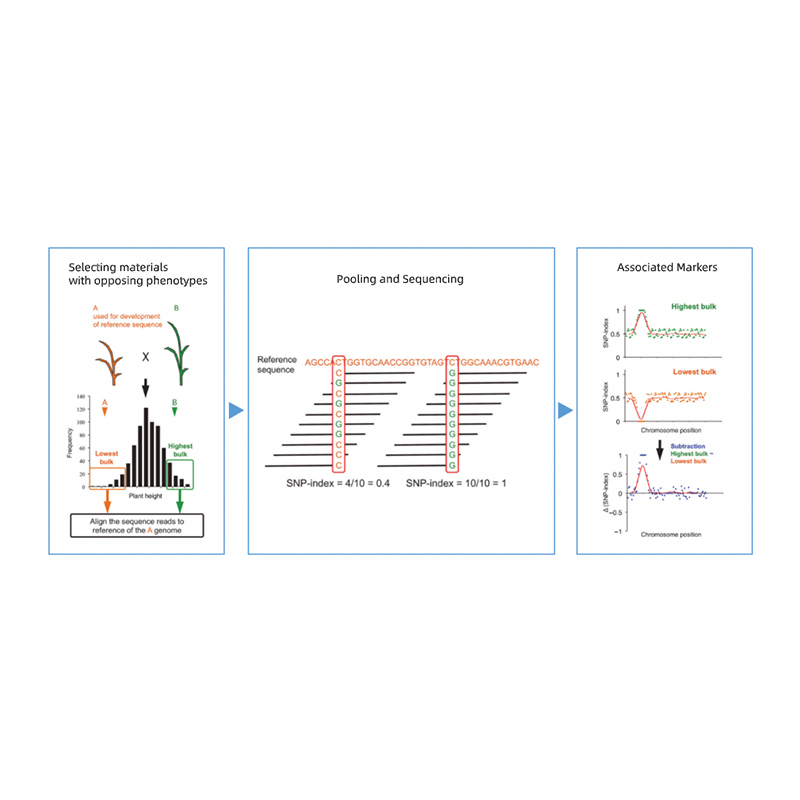
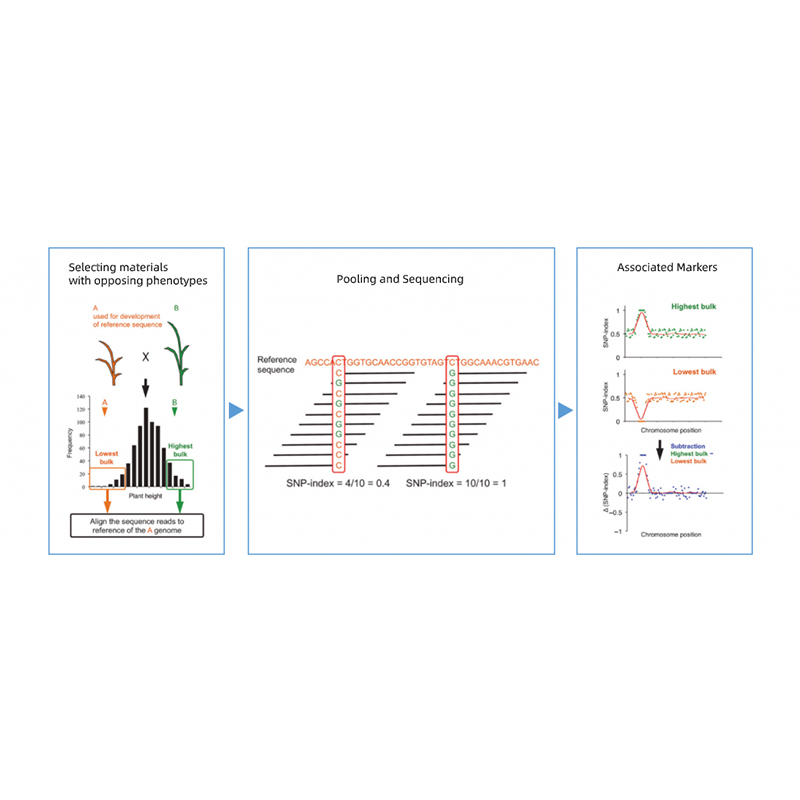
Takagi i in., The Plant Journal, 2013
● Dokładna lokalizacja: mieszanie bali z 30+30 do 200+200 osób w celu zminimalizowania szumu tła; Nie synonimowe prognozy regionu kandydującego oparte na mutatantach.
● Kompleksowa analiza: dogłębna adnotacja funkcji genów kandydujących, w tym NR, SWISSPROT, GO, KEGG, COG, KOG itp.
● Szybszy czas zwrotu: szybka lokalizacja genów w ciągu 45 dni roboczych.
● Rozległe doświadczenie: BMK przyczyniło się do tysięcy lokalizacji cech, obejmując różnorodne gatunki, takie jak uprawy, produkty wodne, las, kwiaty, owoce itp.
Specyfikacje usług
Populacja:
Segregowanie potomstwa rodziców z przeciwnymi fenotypami.
EG F2 potomstwo, krzyżowanie wsteczne (BC), rekombinowana linia wsobkowa (RIL)
Mieszanie puli
Dla cech jakościowych: od 30 do 50 osób (minimum 20)/masa
Dla ilościowych Tratis: od 5% do 10% osób z ekstremalnymi fenotypami w całej populacji (minimum 30+30).
Zalecana głębokość sekwencjonowania
Co najmniej 20x/rodzic i 1x/potomstwo (np. Dla puli mieszania potomstwa 30+30 indywidualnych, głębokość sekwencjonowania będzie wynosić 30x na luzem)
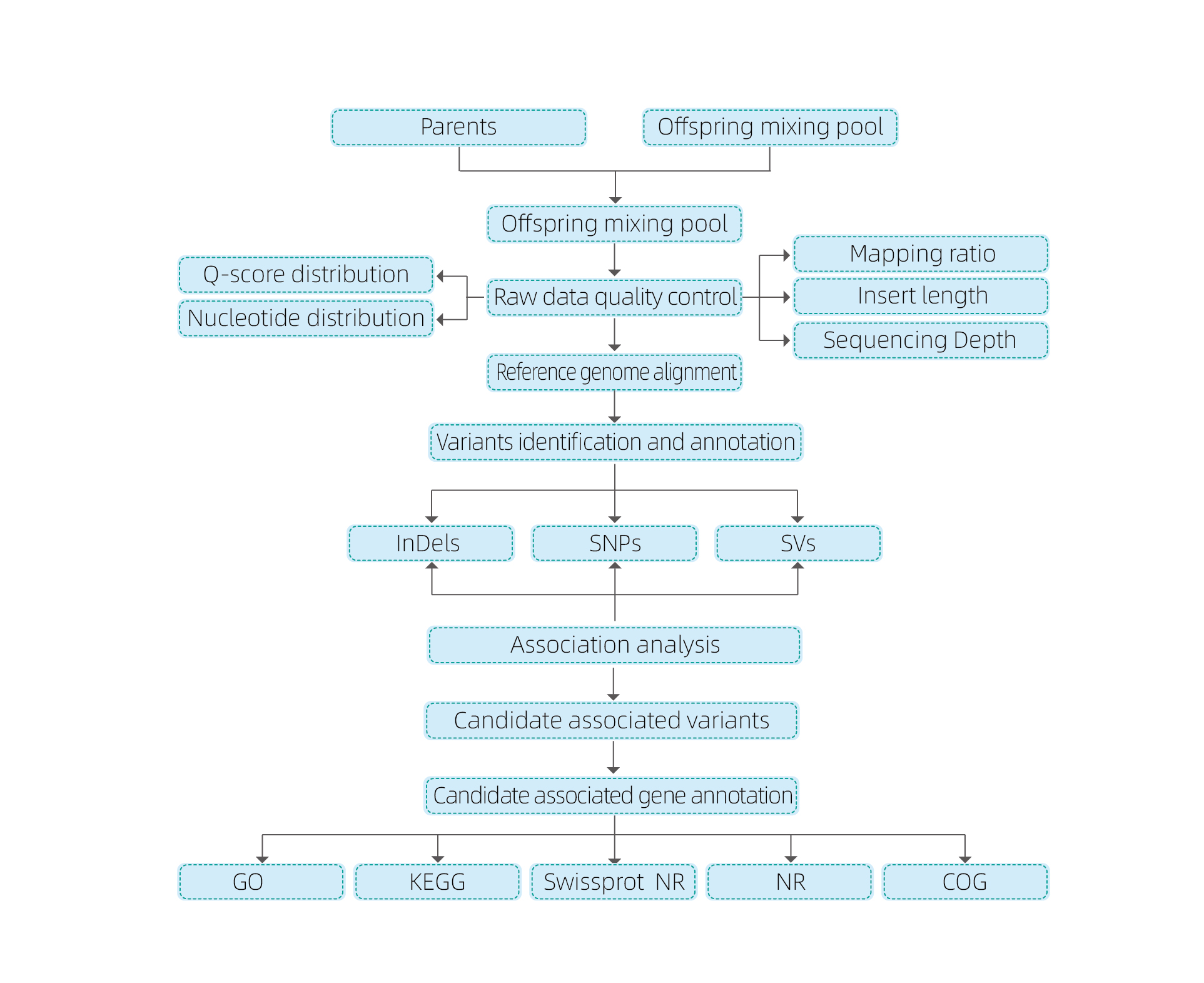
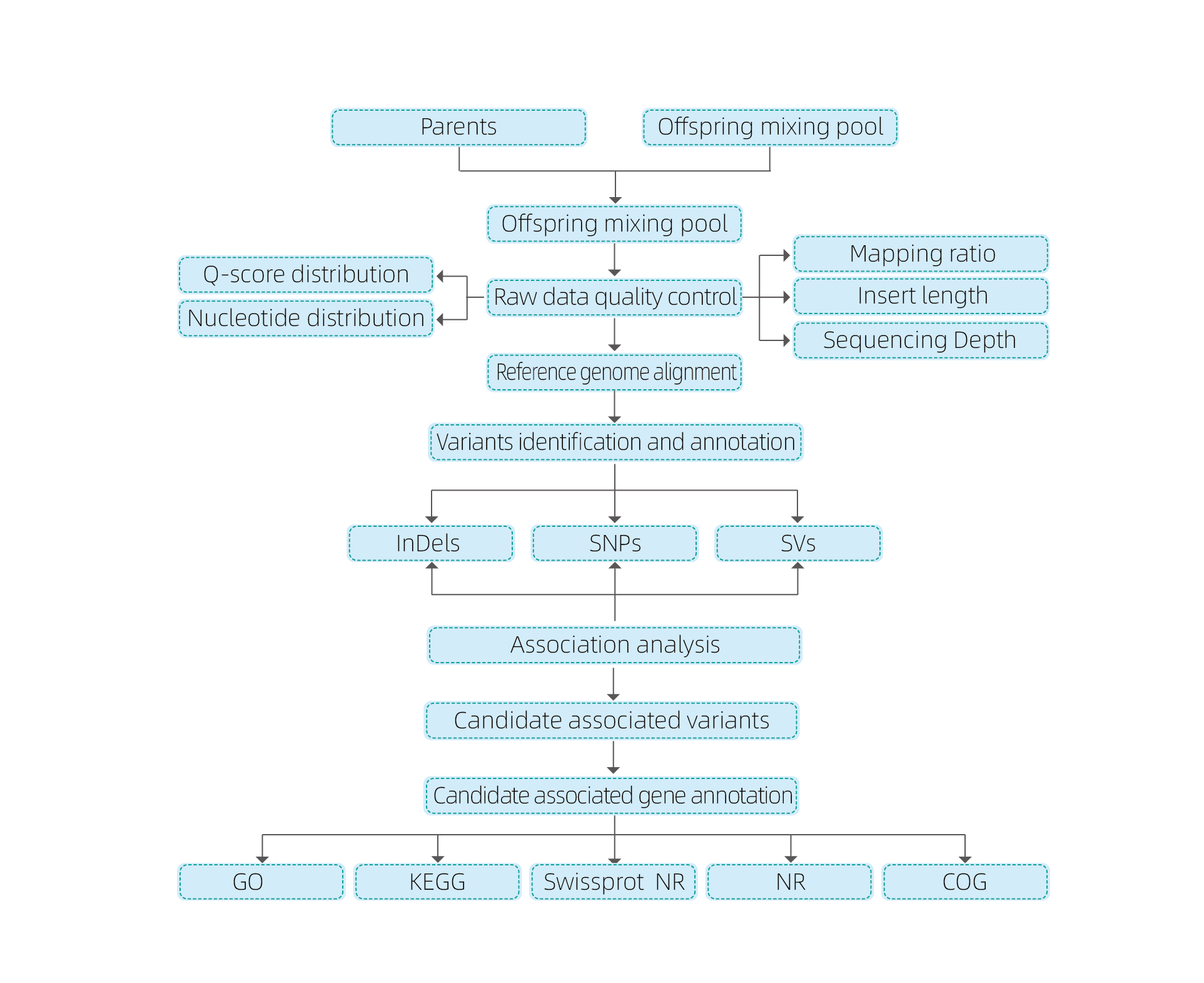
Analizy bioinformatyczne
● Resekwencjonowanie całego genomu
● Przetwarzanie danych
● Połączenie SNP/Indel
● Badanie badań regionu kandydującego
● Adnotacja funkcji genów kandydujących
Przykładowe wymagania i dostawa
Przykładowe wymagania:
Nukleotydy:
| Próbka GDNA | Próbka tkanki |
| Stężenie: ≥30 ng/μl | Rośliny: 1-2 g |
| Ilość: ≥2 μg (Volumn ≥15 μl) | Zwierzęta: 0,5-1 g |
| Czystość: OD260/280 = 1,6-2,5 | Krew pełna: 1,5 ml |
Przepływ pracy usług

Projekt eksperymentu

Dostawa próbki

Ekstrakcja RNA

Konstrukcja biblioteki

Sekwencjonowanie

Analiza danych

Usługi po sprzedaży
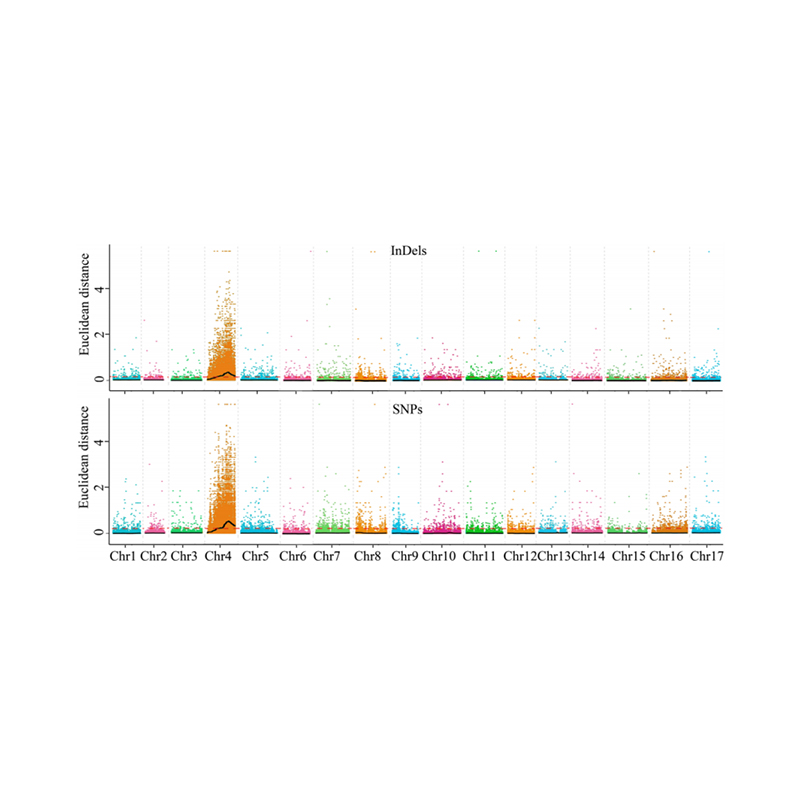
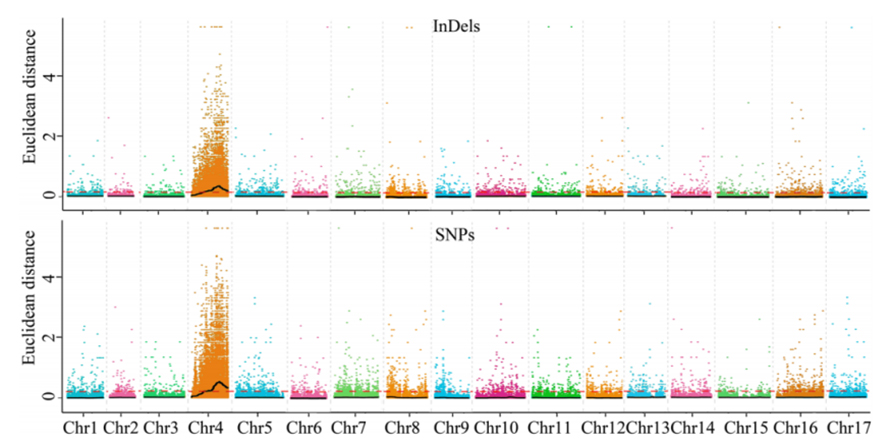
1. Baza analizy asocjacji na odległości euklidesowej (ED) w celu zidentyfikowania regionu kandydującego. Na poniższym rysunku
Oś X: numer chromosomu; Każda kropka reprezentuje wartość ED SNP. Czarna linia odpowiada dopasowanej wartości ED. Wyższa wartość ED wskazuje na bardziej znaczący związek między miejscem a fenotypem. Czerwona linia deski rozdzielczej reprezentuje próg znaczącego powiązania.
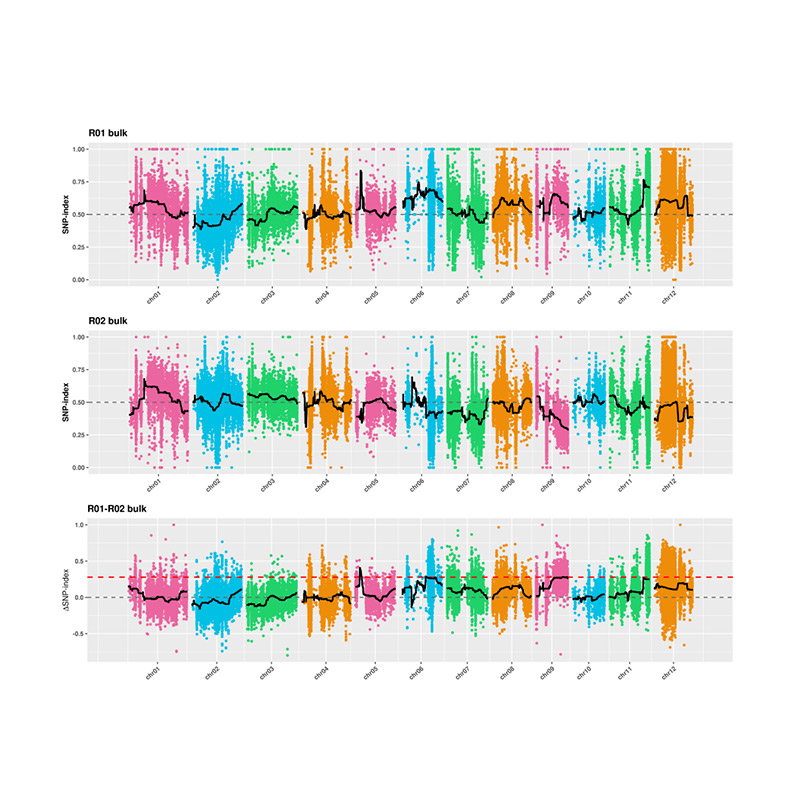
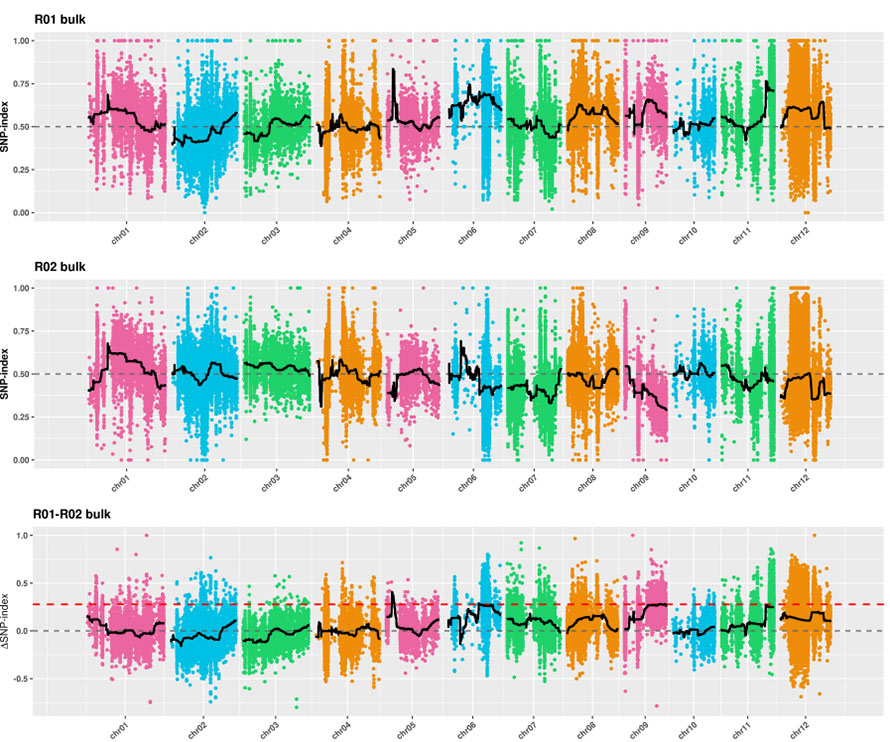
2. Analiza asocjacji bez indeksu SNP
Oś X: numer chromosomu; Każda kropka reprezentuje wartość indeksu SNP. Czarna linia oznacza dopasowaną wartość indeksu SNP. Im większa jest wartość, tym bardziej znaczące jest powiązanie.
Przypadek BMK
Główny wpływ ilościowy locus fnl7.1 koduje późną embriogenezę obfite białko związane z długością owoców szyi w ogórku
Opublikowany: Plant Biotechnology Journal, 2020
Strategia sekwencjonowania:
Rodzice (Jin5-508, YN): Resekwencjonowanie całego genomu dla 34 × i 20 ×.
Baseny DNA (50 długoterminowych i 50 krótkich szyi): resekwencja dla 61 × i 52 ×
Kluczowe wyniki
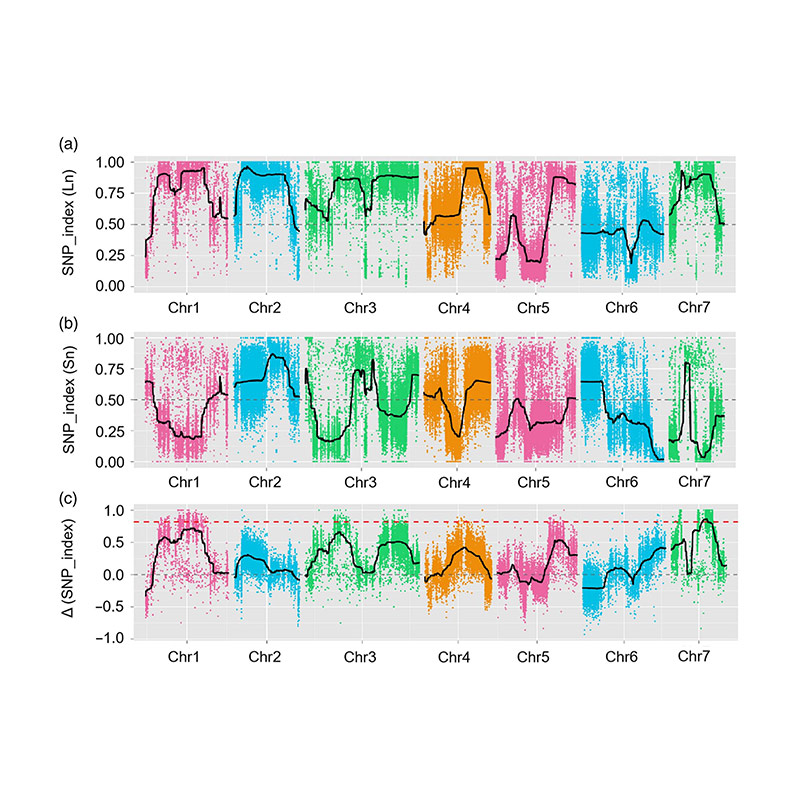
W tym badaniu wygenerowano segregację populacji (F2 i F2: 3) przez przekroczenie linii ogórka ogórków z długiej szyi JIN5-508 i YN z krótkim szyją. Dwie pule DNA zostały zbudowane przez 50 ekstremalnych osób z długim dekoltem i 50 ekstremalnych osobników z krótkim drzemką. QTL znaczącego efektu zidentyfikowano na podstawie Analizy BSA i tradycyjnym mapowaniu QTL. Region kandydujący został dalej zmniejszony przez drobne mapowanie, kwantyfikację ekspresji genów i eksperymenty transgeniczne, które ujawniły kluczowy gen w kontrolowaniu długości szyi, CSFNL7.1. Ponadto stwierdzono, że polimorfizm w regionie promotora CSFNL7.1 jest związany z odpowiadającym wyrażeniem. Dalsza analiza filogenetyczna sugerowała, że locus FNL7.1 jest bardzo prawdopodobny z Indii.
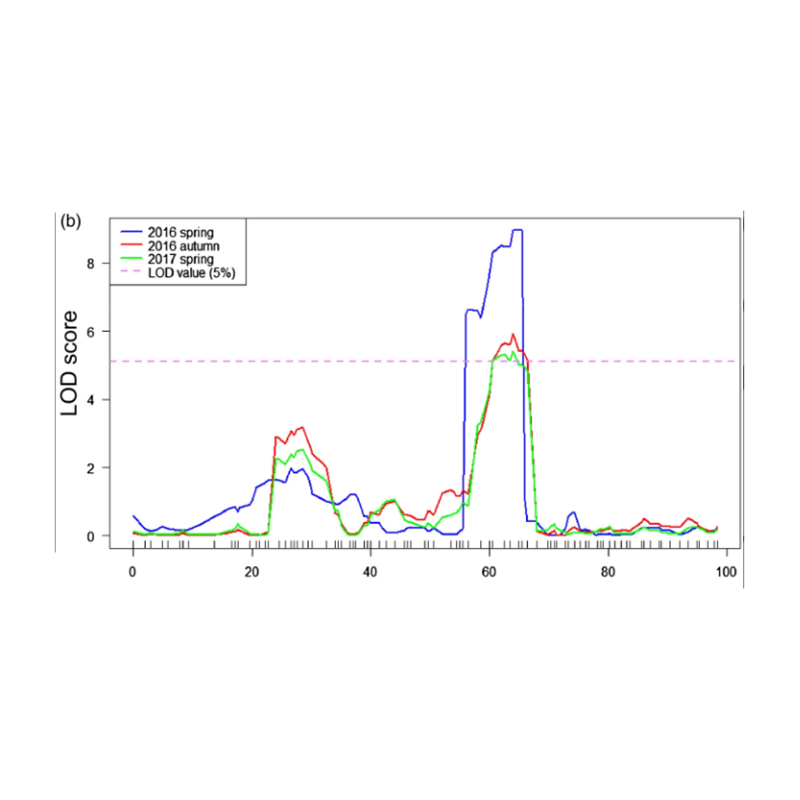
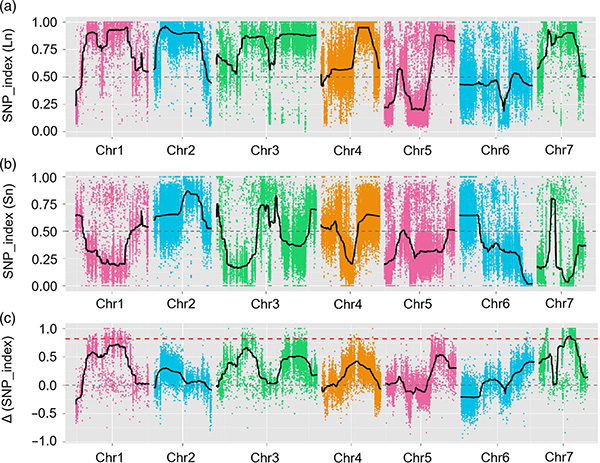 Mapowanie QTL w analizie BSA w celu zidentyfikowania regionu kandydującego związanego z długością szyi ogórka | 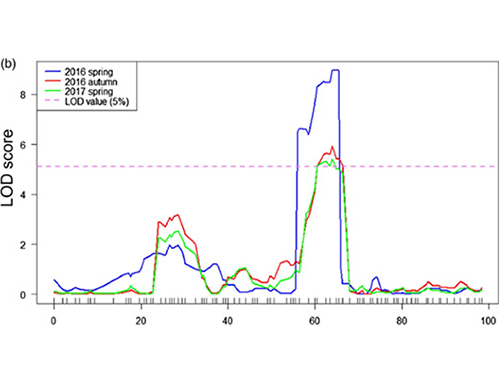 Profile LOD QTL o długości szyi ogórkowej zidentyfikowane na CHR07 |
Xu, X. i in. „Główny efekt ilościowy locus FNL7.1 koduje późną embriogenezę obfite białko związane z długością szyi owocowej w ogórku”. Plant Biotechnology Journal 18.7 (2020).



