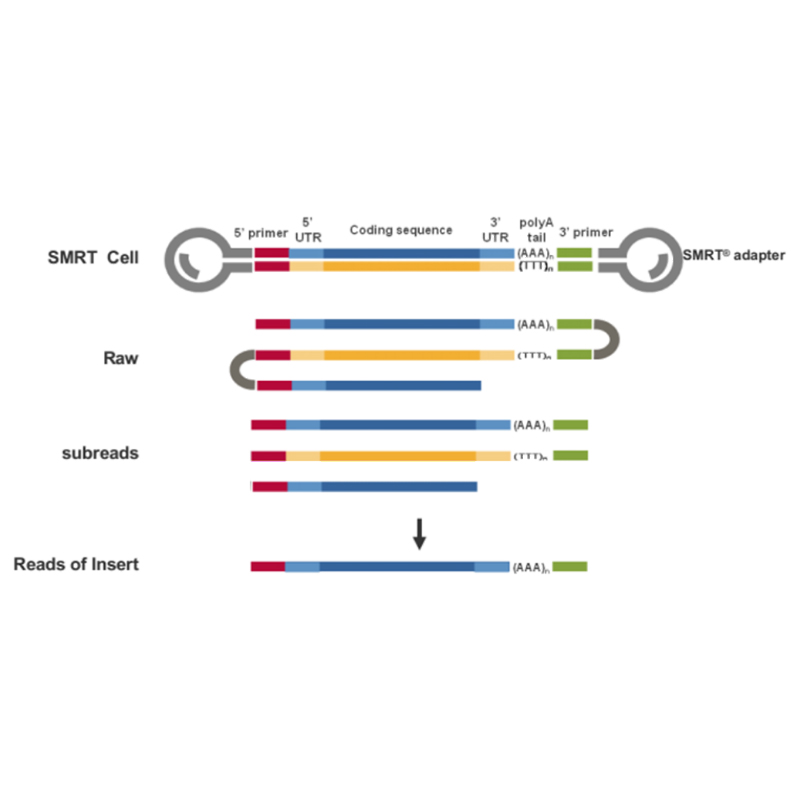
ການຈັດລຳດັບ mRNA ເຕັມຄວາມຍາວ -PacBio

ຄຸນສົມບັດ
● ການສັງເຄາະ cDNA ຈາກ poly-A mRNA ຕາມດ້ວຍການກະກຽມຫ້ອງສະໝຸດ
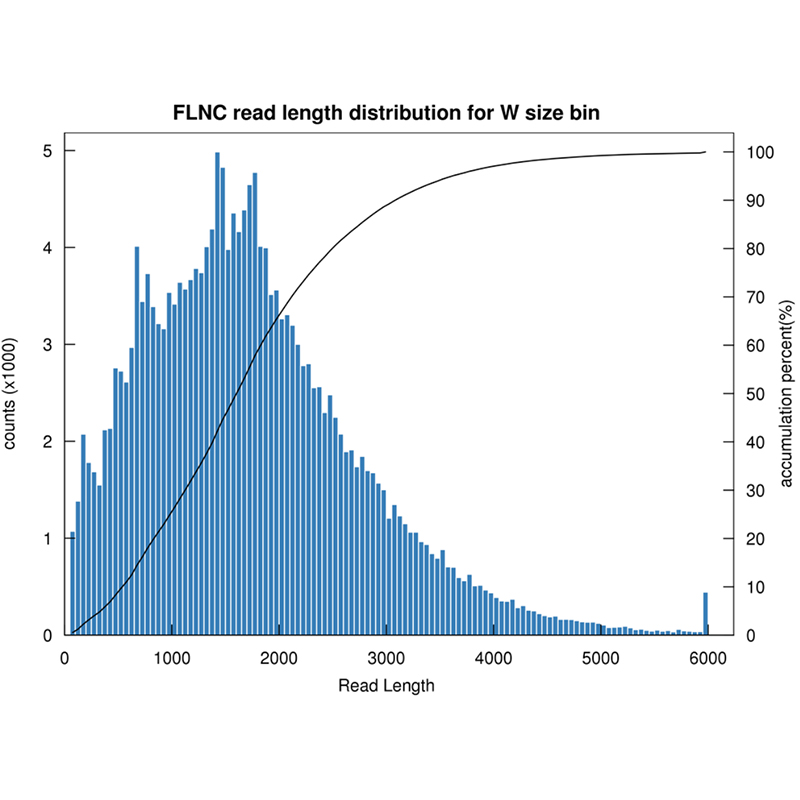
● ການຈັດລໍາດັບໃນໂໝດ CCS, ສ້າງການອ່ານ HiFi
● ການຈັດລໍາດັບຂອງຂໍ້ຄວາມຖອດຈາກສຽງສະບັບເຕັມ
● ການວິເຄາະບໍ່ຈໍາເປັນຕ້ອງມີ genome ອ້າງອີງ; ຢ່າງໃດກໍຕາມ, ມັນອາດຈະໄດ້ຮັບການຈ້າງງານ
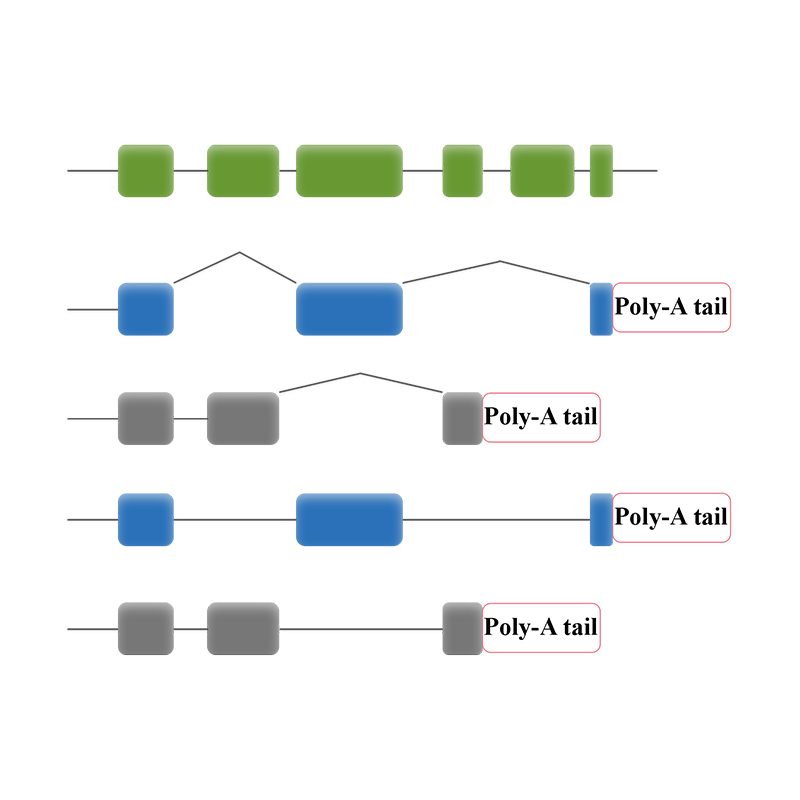
● ການວິເຄາະທາງຊີວະພາບເຮັດໃຫ້ການວິເຄາະຂໍ້ມູນການຖອດຂໍ້ຄວາມ isoform lncRNA, gene fusions, poly-adenylation, ແລະໂຄງສ້າງຂອງ gene
ຂໍ້ໄດ້ປຽບການບໍລິການ
●ຄວາມຖືກຕ້ອງສູງ: HiFi ອ່ານດ້ວຍຄວາມຖືກຕ້ອງ > 99.9% (Q30), ທຽບກັບ NGS
● ການວິເຄາະ Splicing ທາງເລືອກ: ການຈັດລຽງລຳດັບຂອງບົດບັນທຶກທັງໝົດເຮັດໃຫ້ການລະບຸຕົວຕົນ ແລະລັກສະນະ isoform
●ຊ່ຽວຊານຢ່າງກວ້າງຂວາງ: ດ້ວຍບັນທຶກການຕິດຕາມຂອງການເຮັດສໍາເລັດຫຼາຍກວ່າ 1100 PacBio ໂຄງການ transcriptome ທີ່ມີຄວາມຍາວເຕັມແລະການປຸງແຕ່ງຫຼາຍກວ່າ 2300 ຕົວຢ່າງ, ທີມງານຂອງພວກເຮົານໍາເອົາປະສົບການທີ່ອຸດົມສົມບູນໃຫ້ກັບທຸກໆໂຄງການ.
●ສະຫນັບສະຫນູນການຂາຍຫລັງ: ຄໍາຫມັ້ນສັນຍາຂອງພວກເຮົາຂະຫຍາຍອອກໄປນອກເຫນືອຈາກການສໍາເລັດໂຄງການທີ່ມີໄລຍະເວລາການບໍລິການຫລັງການຂາຍ 3 ເດືອນ. ໃນລະຫວ່າງເວລານີ້, ພວກເຮົາສະເຫນີການຕິດຕາມໂຄງການ, ການຊ່ວຍເຫຼືອການແກ້ໄຂບັນຫາ, ແລະກອງປະຊຸມ Q&A ເພື່ອແກ້ໄຂຄໍາຖາມໃດໆທີ່ກ່ຽວຂ້ອງກັບຜົນໄດ້ຮັບ.
ຄວາມຕ້ອງການຕົວຢ່າງແລະການຈັດສົ່ງ
| ຫໍສະໝຸດ | ຍຸດທະສາດການຈັດລໍາດັບ | ຂໍ້ມູນແນະນໍາ | ການຄວບຄຸມຄຸນນະພາບ |
| ຫ້ອງສະໝຸດ mRNA CCS ທີ່ອຸດົມໄປດ້ວຍ PolyA | PacBio Sequel II PacBio Revio | 20/40 Gb 5/10 M CCS | Q30≥85% |
ຄວາມຕ້ອງການຕົວຢ່າງ:
Nucleotides:
● ພືດ:
ຮາກ, ລຳຕົ້ນ ຫຼື ກີບດອກ: 450 ມກ
ໃບ ຫຼື ແກ່ນ: 300 ມກ
ໝາກ: 1.2 g
● ສັດ:
ຫົວໃຈ ຫຼື ລຳໄສ້: 300 ມກ
Viscera ຫຼືສະຫມອງ: 240 ມກ
ກ້າມເນື້ອ: 450 ມກ
ກະດູກ, ຜົມ ຫຼື ຜິວໜັງ: 1g
● Arthropods:
ແມງໄມ້: 6g
Crustacea: 300 ມກ
● ເລືອດທັງໝົດ: 1 ທໍ່
● ເຊລ: 106 ຈຸລັງ
| Conc.(ng/μl) | ປະລິມານ (μg) | ຄວາມບໍລິສຸດ | ຄວາມຊື່ສັດ |
| ≥ 100 | ≥ 1.0 | OD260/280=1.7-2.5 OD260/230=0.5-2.5 ຈໍາກັດຫຼືບໍ່ມີການປົນເປື້ອນທາດໂປຼຕີນຫຼື DNA ທີ່ສະແດງຢູ່ໃນເຈນ. | ສໍາລັບພືດ: RIN≥7.5; ສໍາລັບສັດ: RIN≥8.0; 5.0≥ 28S/18S≥1.0; ຂອບເຂດຈໍາກັດ ຫຼືບໍ່ມີລະດັບຄວາມສູງ |
ການຈັດສົ່ງຕົວຢ່າງທີ່ແນະນໍາ
ຕູ້ຄອນເທນເນີ: ທໍ່ centrifuge 2 ມລ (ບໍ່ແນະນໍາໃຫ້ໃຊ້ຟອຍກົ່ວ)
ການຕິດສະຫຼາກຕົວຢ່າງ: Group+replicate ເຊັ່ນ: A1, A2, A3; B1, B2, B3.
ການຂົນສົ່ງ:
1. ນ້ຳກ້ອນແຫ້ງ: ຕົວຢ່າງຕ້ອງຖືກບັນຈຸໃສ່ຖົງ ແລະຝັງໄວ້ໃນນ້ຳກ້ອນແຫ້ງ.
2. ທໍ່ RNAstable: ຕົວຢ່າງ RNA ສາມາດຕາກແຫ້ງໃນທໍ່ສະຖຽນລະພາບ RNA (ເຊັ່ນ: RNAstable®) ແລະສົ່ງໃນອຸນຫະພູມຫ້ອງ.
ກະແສວຽກບໍລິການ

ການອອກແບບທົດລອງ

ການຈັດສົ່ງຕົວຢ່າງ

ການສະກັດເອົາ RNA

ການກໍ່ສ້າງຫໍສະຫມຸດ

ການຈັດລໍາດັບ

ການວິເຄາະຂໍ້ມູນ

ບໍລິການຫຼັງການຂາຍ

ລວມມີການວິເຄາະຕໍ່ໄປນີ້:
● ການຄວບຄຸມຄຸນນະພາບຂໍ້ມູນດິບ
● ການວິເຄາະ Polyadenylation ທາງເລືອກ (APA)
● ການວິເຄາະການຖອດຂໍ້ຄວາມແບບຟິວຊັນ
● ການວິເຄາະ Splicing ທາງເລືອກ
● Benchmarking Universal Single-Copy Orthlogs (BUSCO) ການວິເຄາະ
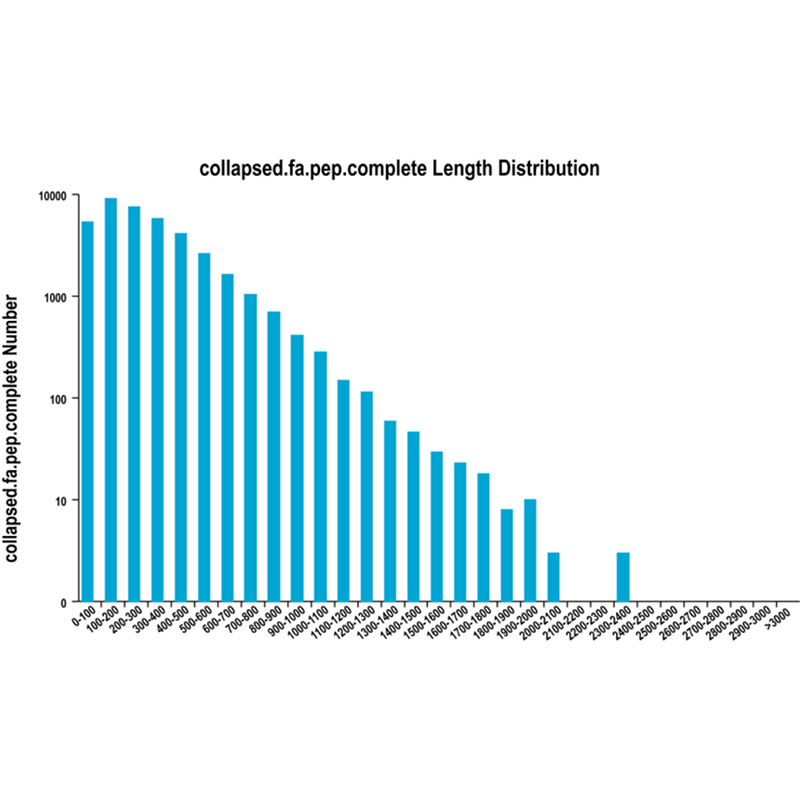
● ການວິເຄາະການຖອດຂໍ້ຄວາມແບບໃໝ່: ການຄາດຄະເນຂອງລຳດັບການເຂົ້າລະຫັດ (CDS) ແລະຄຳອະທິບາຍທີ່ເປັນປະໂຫຍດ
● ການວິເຄາະ lncRNA: ການຄາດຄະເນຂອງ lncRNA ແລະເປົ້າຫມາຍ
● ການລະບຸຕົວຕົນຂອງ MicroSatelite (SSR)
ການວິເຄາະ BUSCO

ການວິເຄາະ Splicing ທາງເລືອກ

ການວິເຄາະ Polyadenylation ທາງເລືອກ (APA)

ຄຳອະທິບາຍປະກອບທີ່ມີປະໂຫຍດຂອງການຖອດຂໍ້ຄວາມໃໝ່

ສຳຫຼວດຄວາມກ້າວໜ້າທີ່ອຳນວຍຄວາມສະດວກໂດຍການບໍລິການຈັດລຽງລຳດັບ mRNA ເຕັມຄວາມຍາວຂອງ BMKGene ໃນສິ່ງພິມທີ່ໂດດເດັ່ນນີ້.
Ma, Y. et al. (2023) 'ການວິເຄາະປຽບທຽບຂອງ PacBio ແລະ ONT RNA ວິທີການຈັດລໍາດັບສໍາລັບ Nemopilema Nomurai venom identification', Genomics, 115(6), p. 110709. doi: 10.1016/J.YGENO.2023.110709.
Chao, Q. et al. (2019) 'ນະໂຍບາຍດ້ານການພັດທະນາຂອງ stem transcriptome', Plant Biotechnology Journal, 17(1), ໜ້າ 206–219. doi: 10.1111/PBI.12958.
Deng, H. et al. (2022) 'ການປ່ຽນແປງແບບເຄື່ອນໄຫວຂອງເນື້ອໃນອາຊິດ Ascorbic ໃນລະຫວ່າງການພັດທະນາໝາກ ແລະ ການສຸກຂອງໝາກ Actinidia latifolia (ການປູກພືດໝາກໄມ້ທີ່ອຸດົມສົມບູນ Ascorbate) ແລະ ກົນໄກໂມເລກຸນທີ່ກ່ຽວຂ້ອງ', International Journal of Molecular Sciences, 23(10), ໜ້າ. 5808. doi: 10.3390/IJMS23105808/S1.
Hua, X. et al. (2022) 'ການຄາດເດົາປະສິດທິພາບຂອງພັນທຸກໍາທາງຊີວະວິທະຍາທີ່ກ່ຽວຂ້ອງກັບໂພລີຟີລິນທາງຊີວະພາບໃນປາຣີໂພລີຟີລາ', ຊີວະວິທະຍາການສື່ສານ 2022 5:1, 5(1), ໜ້າ 1-10. doi: 10.1038/s42003-022-03000-z.
Liu, M. et al. (2023) 'ການປະສົມ PacBio Iso-Seq ແລະ Illumina RNA-Seq ການວິເຄາະຂອງ Tuta absoluta (Meyrick) Transcriptome ແລະ Cytochrome P450 Genes', ແມງໄມ້, 14(4), p. 363. doi: 10.3390/INSECTS14040363/S1.
Wang, Lijun et al. (2019) 'ການສຳຫຼວດຄວາມຊັບຊ້ອນການຖອດຂໍ້ຄວາມໂດຍໃຊ້ PacBio ການວິເຄາະແບບສົດໆໂມເລກຸນດຽວກັບການຈັດລຳດັບ Illumina RNA ເພື່ອຄວາມເຂົ້າໃຈທີ່ດີຂຶ້ນຂອງຊີວະສັງເຄາະອາຊິດ ricinoleic ໃນ Ricinus communis', BMC Genomics, 20(1), ໜ້າ 1-17. doi: 10.1186/S12864-019-5832-9.