

Sequenziamento di frammenti amplificati con locus specifico (SLAF-Seq)

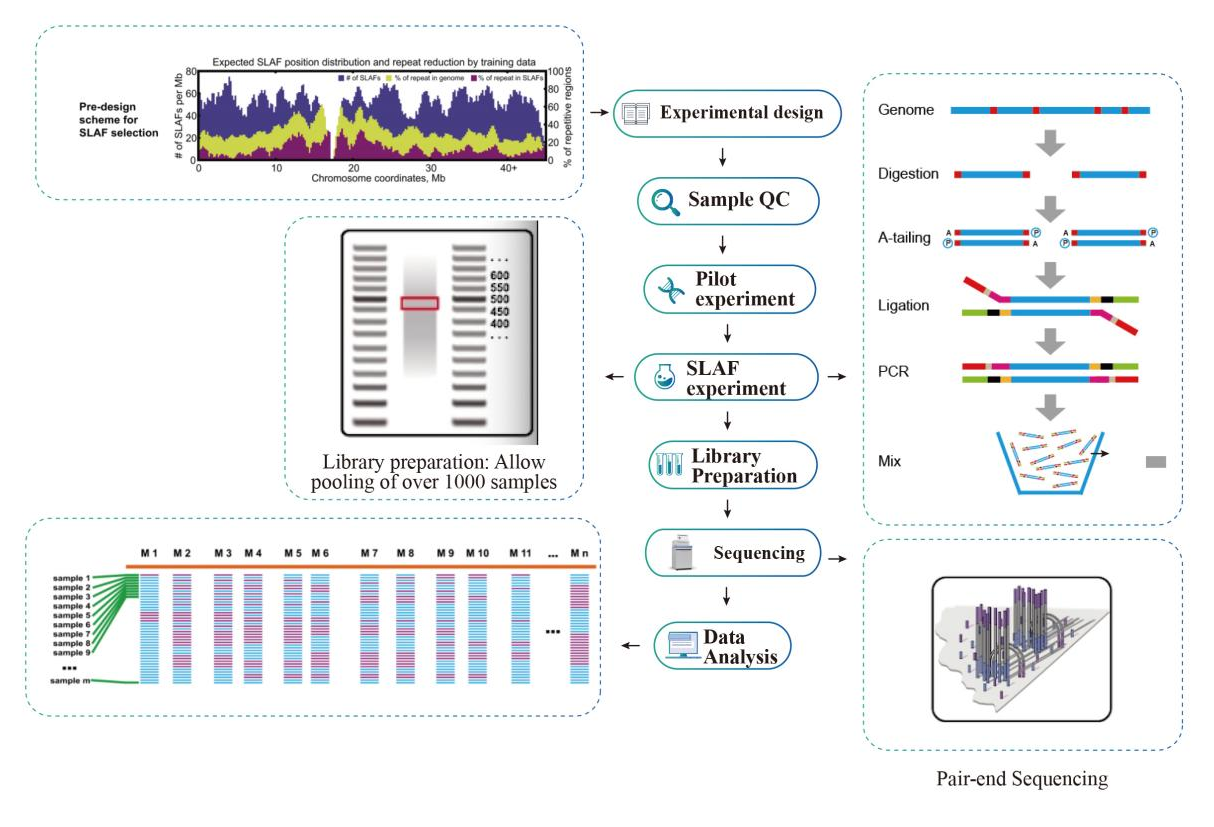
Flusso di lavoro

Schema tecnico

Caratteristiche del servizio
● Sequenziamento su NovaSeq con PE150.
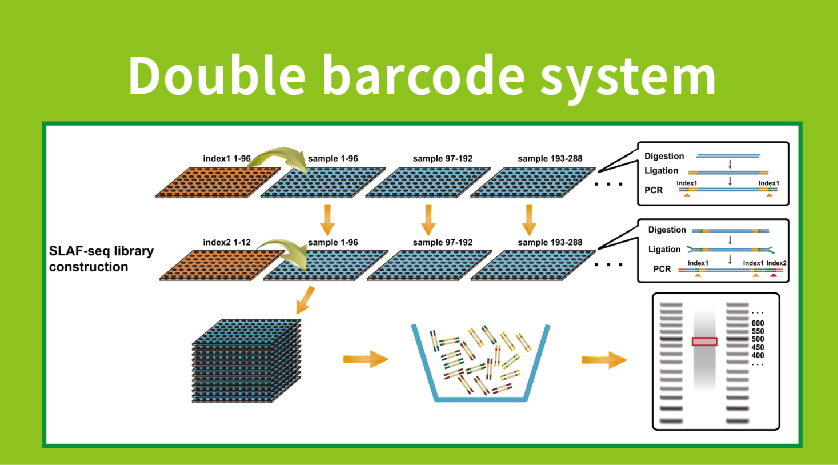
● Preparazione delle librerie con doppio codice a barre, che consente il raggruppamento di oltre 1.000 campioni.
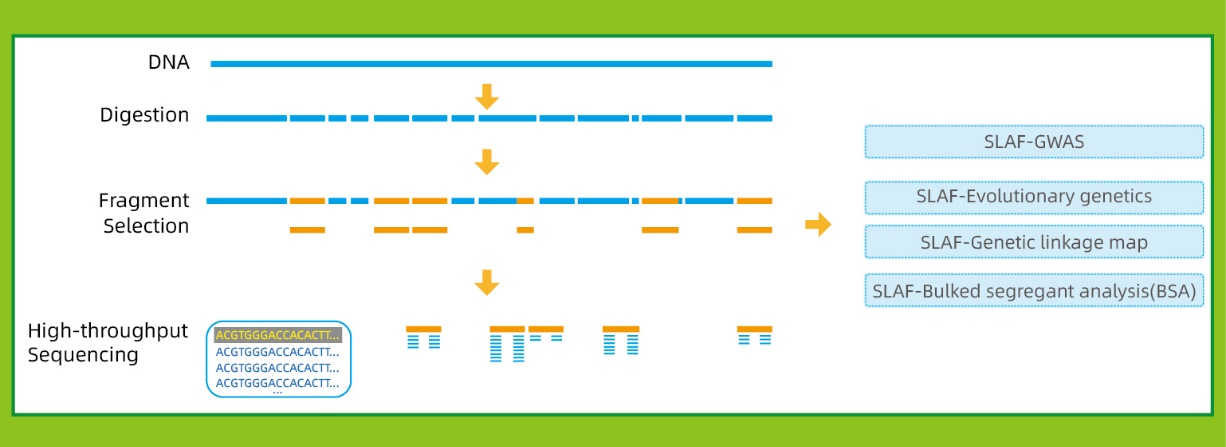
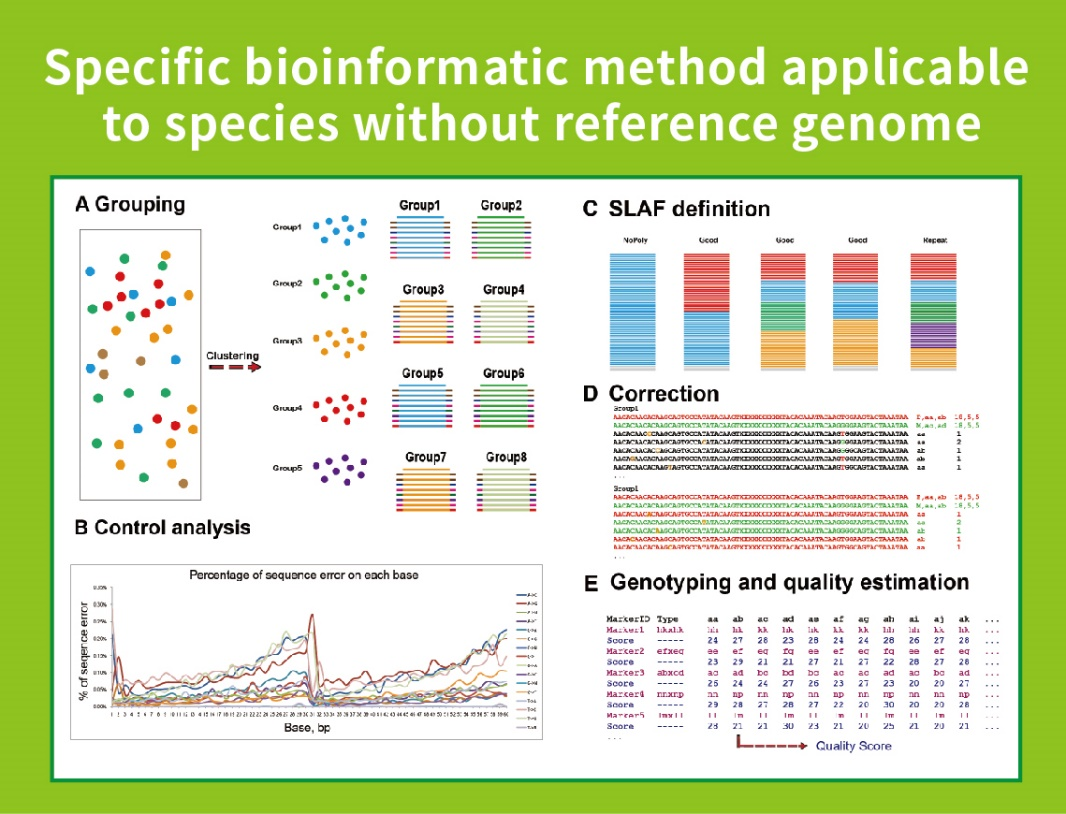
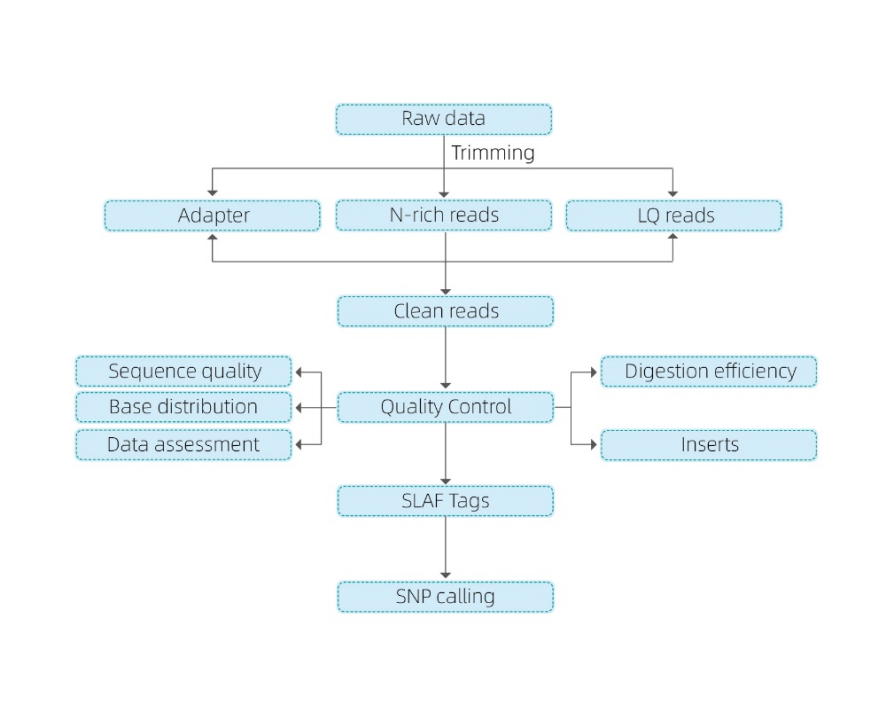
● Questa tecnica può essere utilizzata con o senza genoma di riferimento, con diverse pipeline bioinformatiche per ciascun caso:
Con genoma di riferimento: scoperta SNP e InDel
Senza genoma di riferimento: clustering dei campioni e scoperta di SNP
● Nelin siliconella fase di pre-progettazione vengono selezionate combinazioni multiple di enzimi di restrizione per trovare quelle che generano una distribuzione uniforme di tag SLAF lungo il genoma.
● Durante il pre-esperimento, tre combinazioni di enzimi vengono testate in 3 campioni per generare 9 librerie SLAF e queste informazioni vengono utilizzate per scegliere la combinazione di enzimi di restrizione ottimale per il progetto.
Vantaggi del servizio
●Scoperta di marcatori genetici elevati: L'integrazione di un sistema a doppio codice a barre ad alta produttività consente il sequenziamento simultaneo di grandi popolazioni e l'amplificazione specifica del locus migliora l'efficienza, garantendo che i numeri dei tag soddisfino i diversi requisiti di varie domande di ricerca.
● Bassa dipendenza dal genoma: Può essere applicato a specie con o senza genoma di riferimento.
●Progettazione di schemi flessibili: È possibile selezionare la digestione a singolo enzima, a doppio enzima, a multienzima e vari tipi di enzimi per soddisfare diversi obiettivi di ricerca o specie. ILin silicoviene effettuata una pre-progettazione per garantire una progettazione ottimale dell'enzima.
● Alta efficienza nella digestione enzimatica: La conduzione di unin silicola pre-progettazione e un pre-esperimento hanno assicurato una progettazione ottimale con distribuzione uniforme dei tag SLAF sul cromosoma (1 tag SLAF/4Kb) e sequenza ripetitiva ridotta (<5%).
●Ampia competenza: Il nostro team apporta una vasta esperienza a ogni progetto, con un track record di chiusura di oltre 5000 progetti SLAF-Seq su centinaia di specie, tra cui piante, mammiferi, uccelli, insetti e organismi acquatici.
● Flusso di lavoro bioinformatico autosviluppato: BMKGENE ha sviluppato un flusso di lavoro bioinformatico integrato per SLAF-Seq per garantire l'affidabilità e l'accuratezza dell'output finale.
Specifiche del servizio
| Tipo di analisi | Scala di popolazione consigliata | Strategia di sequenziamento | |
| Profondità di sequenziamento dei tag | Numero dell'etichetta | ||
| Mappe genetiche | 2 genitori e >150 figli | Genitori: 20x WGS Distribuzione: 10x | Dimensione del genoma: <400 Mb: si consiglia WGS <1 Gb: 100.000 tag 1-2Gb:: 200.000 tag >2Gb: 300.000 tag Massimo 500.000 tag |
| Studi di associazione su tutto il genoma (GWAS) | ≥200 campioni | 10x | |
| Evoluzione genetica | ≥30 campioni, con >10 campioni da ciascun sottogruppo | 10x | |
Requisiti del servizio
Concentrazione ≥ 5 ng/μL
Quantità totale ≥ 80 ng
Nanogoccia OD260/280=1,6-2,5
Gel di agarosio: degradazione o contaminazione assente o limitata
Consegna del campione consigliata
Contenitore: provetta da centrifuga da 2 ml
(Per la maggior parte dei campioni si consiglia di non conservare in etanolo)
Etichettatura dei campioni: i campioni devono essere chiaramente etichettati e identici al modulo informativo del campione inviato.
Spedizione: Ghiaccio secco: i campioni devono essere prima imballati in sacchetti e sepolti nel ghiaccio secco.
Flusso di lavoro del servizio






Controllo qualità del campione
Esperimento pilota
Esperimento SLAF
Preparazione della biblioteca
Sequenziamento
Analisi dei dati
Servizi post-vendita
 Include la seguente analisi:
Include la seguente analisi:

- QC dei dati di sequenziamento
- Sviluppo tag SLAF
Mappatura al genoma di riferimento
Senza genoma di riferimento: clustering
- Analisi dei tag SLAF.: statistica, distribuzione nel genoma
- Scoperta dei marcatori: SNP, InDel, SNV, chiamate e annotazioni CV
Distribuzione dei tag SLAF sui cromosomi:

Distribuzione degli SNP sui cromosomi:
 Annotazione SNP
Annotazione SNP

| Anno | Diario | IF | Titolo | Applicazioni |
| 2022 | Comunicazioni della natura | 17.694 | Basi genomiche dei giga-cromosomi e giga-genoma della peonia arborea Peonia ostii | SLAF-GWAS |
| 2015 | Nuovo fitologo | 7.433 | Le impronte della domesticazione ancorano le regioni genomiche di importanza agronomica semi di soia | SLAF-GWAS |
| 2022 | Giornale di ricerca avanzata | 12.822 | Introgressioni artificiali su tutto il genoma di Gossypium barbadense in G. hirsutum rivelano loci superiori per il miglioramento simultaneo della qualità e della resa della fibra di cotone tratti | SLAF-Genetica evolutiva |
| 2019 | Pianta molecolare | 10.81 | L'analisi genomica della popolazione e l'assemblaggio de novo rivelano l'origine di Weedy Il riso come gioco evolutivo | SLAF-Genetica evolutiva |
| 2019 | Genetica della natura | 31.616 | Sequenza del genoma e diversità genetica della carpa comune, Cyprinus carpio | Mappa del collegamento SLAF |
| 2014 | Genetica della natura | 25.455 | Il genoma dell'arachide coltivata fornisce informazioni sui cariotipi dei legumi, poliploidi Evoluzione e domesticazione delle colture. | Mappa del collegamento SLAF |
| 2022 | Giornale delle biotecnologie vegetali | 9.803 | L'identificazione di ST1 rivela una selezione che coinvolge l'autostop della morfologia del seme e il contenuto di olio durante la domesticazione della soia | Sviluppo di marcatori SLAF |
| 2022 | Giornale internazionale di scienze molecolari | 6.208 | Identificazione e sviluppo di marcatori di DNA per un Wheat-Leymus mollis 2Ns (2D) Sostituzione cromosomica disomica | Sviluppo di marcatori SLAF |
| Anno | Diario | IF | Titolo | Applicazioni |
| 2023 | Frontiere nella scienza delle piante | 6.735 | Mappatura QTL e analisi del trascrittoma del contenuto di zucchero durante la maturazione del frutto di Pyrus pyrifolia | Mappa genetica |
| 2022 | Giornale delle biotecnologie vegetali | 8.154 | L'identificazione di ST1 rivela una selezione che coinvolge l'autostop della morfologia dei semi e del contenuto di olio durante la domesticazione della soia
| Chiamata SNP |
| 2022 | Frontiere nella scienza delle piante | 6.623 | Mappatura dell'associazione a livello genomico di fenotipi appena senza scafo in ambienti siccitosi.
| GWAS |





