

Sequenziamento del genoma de novo di piante/animali





De Novoil sequenziamento si riferisce alla costruzione dell'intero genoma di una specie utilizzando tecnologie di sequenziamento in assenza di un genoma di riferimento. L'introduzione e l'adozione diffusa del sequenziamento di terza generazione, caratterizzato da letture più lunghe, hanno migliorato significativamente l'assemblaggio del genoma aumentando la sovrapposizione tra le letture. Questo miglioramento è particolarmente pertinente quando si ha a che fare con genomi complessi, come quelli che presentano un'elevata eterozigosità, un elevato rapporto di regioni ripetitive, poliploidi e regioni con elementi ripetitivi, contenuti GC anomali o elevata complessità che in genere sono scarsamente assemblati utilizzando il sequenziamento a lettura breve solo.
La nostra soluzione completa fornisce servizi di sequenziamento integrati e analisi bioinformatiche che forniscono un genoma assemblato de novo di alta qualità. Un'indagine iniziale del genoma con Illumina fornisce stime delle dimensioni e della complessità del genoma e queste informazioni vengono utilizzate per guidare la fase successiva del sequenziamento a lunga lettura con PacBio HiFi, seguito dade novoassemblaggio di contig. Il successivo utilizzo dell'assemblaggio HiC consente l'ancoraggio dei contig al genoma, ottenendo un assemblaggio a livello cromosomico. Infine, il genoma viene annotato mediante predizione genetica e sequenziamento dei geni espressi, ricorrendo a trascrittomi con letture brevi e lunghe.
Caratteristiche del servizio
● Integrazione di più servizi di sequenziamento e bioinformatici in un'unica soluzione:
Indagine sul genoma con Illumina per stimare la dimensione del genoma e guidare i passaggi successivi;
Sequenza di lettura lunga perde novoassemblaggio di contig;
Sequenziamento Hi-C per l'ancoraggio cromosomico;
sequenziamento dell'mRNA per l'annotazione dei geni;
Validazione dell'assemblea.
● Servizio adatto alla costruzione di nuovi genomi o al miglioramento di genomi di riferimento esistenti per specie di interesse.
Vantaggi del servizio
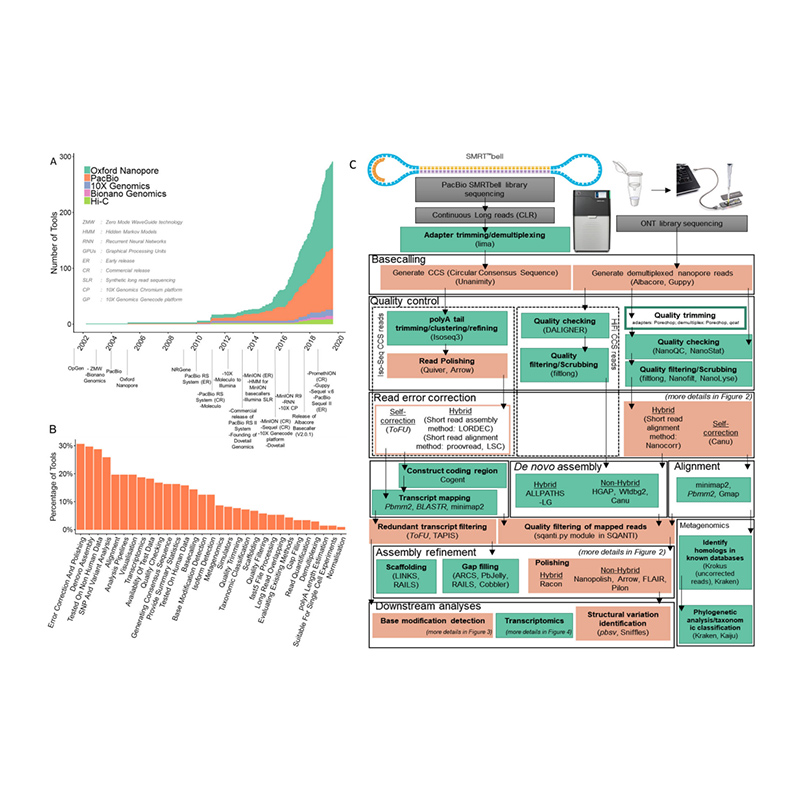
Sviluppo di piattaforme di sequenziamento e bioinformatica inde novoassemblaggio del genoma
(Amarasinghe SL et al.,Biologia del genoma, 2020)
●Vasta esperienza e record di pubblicazioni: BMKGene ha accumulato una vasta esperienza nell'assemblaggio di genomi di alta qualità di diverse specie, inclusi genomi diploidi e genomi altamente complessi di specie poliploidi e allopoliploidi. Dal 2018 abbiamo contribuito a oltre300 pubblicazioni di grande impatto e oltre 20 di queste sono pubblicate su Nature Genetics.
● Soluzione unica: il nostro approccio integrato combina più tecnologie di sequenziamento e analisi bioinformatiche in un flusso di lavoro coerente, offrendo un genoma assemblato di alta qualità.
●Su misura per le tue esigenze: Il flusso di lavoro del nostro servizio è personalizzabile e consente l'adattamento a genomi con caratteristiche diverse ed esigenze di ricerca specifiche. Ciò include l'adattamento di genomi giganti, genomi poliploidi, genomi altamente eterozigoti e altro ancora.
●Team di Bioinformatica e Laboratorio altamente qualificato: con grande esperienza sia sul fronte sperimentale che bioinformatico di assemblaggi genomici complessi e una serie di brevetti e copyright di software.
●Supporto post-vendita:Il nostro impegno si estende oltre il completamento del progetto con un periodo di servizio post-vendita di 3 mesi. Durante questo periodo, offriamo follow-up del progetto, assistenza per la risoluzione dei problemi e sessioni di domande e risposte per rispondere a qualsiasi domanda relativa ai risultati.
Specifiche del servizio
| Indagine sul genoma | Assemblaggio del genoma | A livello cromosomico | Annotazione del genoma |
| 50X Illumina NovaSeq PE150
| Letture HiFi PacBio CCS 30X | 100X Hi-C | RNA-seq Illumina PE150 10 Gb + (opzionale) PacBio RNA-seq a lunghezza intera da 40 Gb o Nanoporo 12 Gb |
Requisiti del servizio
Per Genome Survey, Genome Assembly e Hi-C Assembly:
| Acidi nucleici tissutali o estratti | Indagine sul genoma | Assemblaggio del genoma con PacBio | Assemblea Hi-C |
| Visceri animali | 0,5-1 g
| ≥ 3,5 g | ≥2 g |
| Muscolo animale | ≥ 5 g | ||
| Sangue di mammiferi | 1,5 ml
| ≥ 5ml | ≥2 ml |
| Sangue di pollame/pesce | ≥ 0,5ml | ||
| Pianta: foglia fresca | 1-2 g | ≥ 5 g | ≥ 4 g |
| Cellule in coltura |
| ≥1x108 | ≥1x107 |
| Insetto | 0,5-1 g | ≥ 3 g | ≥ 2 g |
| DNA estratto | Concentrazione: ≥1 ng/μL Quantità ≥ 30 ng Degrado o contaminazione limitati o assenti | Concentrazione: ≥ 50 ng/μL Quantità: 10 µg/cella a flusso/campione DE260/280=1,7-2,2 DE260/230=1,8-2,5 Degrado o contaminazione limitati o assenti |
-
|
Per l'annotazione del genoma con trascrittomica:
| Acidi nucleici tissutali o estratti | Trascrittoma Illumina | Trascrittoma PacBio | Trascrittoma dei nanopori |
| Pianta: radice/stelo/petalo | 450mg | 600 mg | |
| Pianta – Foglia/Seme | 300 mg | 300 mg | |
| Pianta - Frutto | 1,2 g | 1,2 g | |
| Cuore/intestino animale | 300 mg | 300 mg | |
| Visceri/cervello animale | 240 mg | 240 mg | |
| Muscolo animale | 450mg | 450mg | |
| Ossa/peli/pelle di animali | 1 g | 1 g | |
| Artropodi - Insetto | 6 | 6 | |
| Artropodi - Crostacei | 300 mg | 300 mg | |
| Sangue intero | 1 tubo | 1 tubo | |
| RNA estratto | Concentrazione: ≥ 20 ng/μL Quantità ≥ 0,3 µg DE260/280=1,7-2,5 DE260/230=0,5-2,5 RIN≥ 6 5≥28S/18S≥1 | Concentrazione: ≥ 100 ng/μL Quantità ≥ 0,75 µg DE260/280=1,7-2,5 DE260/230=0,5-2,5 RIN≥ 8 5≥28S/18S≥1 | Concentrazione: ≥ 100 ng/μL Quantità ≥ 0,75 µg DE260/280=1,7-2,5 DE260/230=0,5-2,5 RIN≥ 7,5 5≥28S/18S≥1 |
Consegna del campione consigliata
Contenitore: provetta da centrifuga da 2 ml (la carta stagnola non è consigliata)
(Per la maggior parte dei campioni si consiglia di non conservare in etanolo.)
Etichettatura dei campioni: i campioni devono essere chiaramente etichettati e identici al modulo informativo del campione inviato.
Spedizione: Ghiaccio secco: i campioni devono essere prima imballati in sacchetti e sepolti nel ghiaccio secco.
Flusso di lavoro

Flusso di lavoro del servizio

Progettazione dell'esperimento

Consegna del campione

Estrazione del DNA

Costruzione della biblioteca

Sequenziamento

Analisi dei dati

Servizi post-vendita

Analisi bioinformatica completa, divisa in 4 passaggi:
1) Indagine sul genoma, basata sull'analisi k-mer con NGS si legge:
Stima della dimensione del genoma
Stima dell'eterozigosi
Stima delle regioni ripetitive
2) Assemblaggio del genoma con PacBio HiFi:
Di nuovoassemblaggio
Valutazione dell'assemblaggio: inclusa l'analisi BUSCO per la completezza del genoma e la mappatura delle letture NGS e PacBio HiFi
3) Assemblaggio Hi-C:
QC libreria Hi-C: stima delle interazioni Hi-C valide
Assemblaggio Hi-C: raggruppamento di contig in gruppi, seguito dall'ordinamento dei contig all'interno di ciascun gruppo e dall'assegnazione dell'orientamento dei contig
Valutazione Hi-C
4) Annotazione del genoma:
Predizione dell'RNA non codificante
Identificazione di sequenze ripetitive (trasposoni e ripetizioni in tandem)
Previsione genetica
§Di nuovo: algoritmi ab initio
§ Basato sull'omologia
§ Basato sul trascrittoma, con letture lunghe e brevi: le letture sonode novoassemblati o mappati sulla bozza del genoma
§ Annotazione dei geni predetti con più database
1) Genome Survey - analisi k-mer

2) Assemblaggio del genoma

2) Assemblaggio del genoma – PacBio HiFi legge la mappatura nella bozza dell'assemblaggio

2) Assemblaggio Hi-C – stima delle coppie di interazione valide Hi-C

3) Valutazione post-assemblaggio Hi-C

4) Annotazione del genoma – integrazione dei geni previsti

4) Annotazione del genoma – annotazione dei geni previsti

Esplora i progressi facilitati dai servizi di assemblaggio del genoma de novo di BMKGene attraverso una raccolta curata di pubblicazioni:
Li, C. et al. (2021) "Le sequenze del genoma rivelano percorsi di dispersione globale e suggeriscono adattamenti genetici convergenti nell'evoluzione dei cavallucci marini", Nature Communications, 12(1). doi: 10.1038/S41467-021-21379-X.
Li, Y. et al. (2023) "I cambiamenti cromosomici su larga scala portano ad alterazioni dell'espressione a livello del genoma, adattamento ambientale e speciazione nel Gayal (Bos frontalis)", Biologia molecolare ed evoluzione, 40(1). doi: 10.1093/MOLBEV/MSAD006.
Tian, T. et al. (2023) "Assemblaggio del genoma e dissezione genetica di un importante germoplasma di mais resistente alla siccità", Nature Genetics 2023 55:3, 55(3), pp. 496–506. doi: 10.1038/s41588-023-01297-y.
Zhang, F. et al. (2023) "Rivelare l'evoluzione della biosintesi degli alcaloidi tropanici analizzando due genomi nella famiglia delle Solanacee", Nature Communications 2023 14:1, 14(1), pp. 1–18. doi: 10.1038/s41467-023-37133-4.
Casi di studio impegnativi:
Assemblaggio telomero-telomero:Fu, A. et al. (2023) "L'assemblaggio del genoma da telomero a telomero del melone amaro (Momordica charantia L. var. abbreviata Ser.) rivela lo sviluppo del frutto, la composizione e le caratteristiche genetiche della maturazione", Horticulture Research, 10(1). doi: 10.1093/HR/UHAC228.
Assemblaggio dell'aplotipo:Hu, W. et al. (2021) "Il genoma definito dagli alleli rivela la differenziazione biallelica durante l'evoluzione della manioca", Molecular Plant, 14(6), pp. 851–854. doi: 10.1016/j.molp.2021.04.009.
Assemblaggio del genoma gigante:Yuan, J. et al. (2022) "Base genomica dei giga-cromosomi e del giga-genoma della peonia arborea Paeonia ostii", Nature Communications 2022 13:1, 13(1), pp. 1–16. doi: 10.1038/s41467-022-35063-1.
Assemblaggio del genoma poliploide:Zhang, Q. et al. (2022) "Approfondimenti genomici sulla recente riduzione cromosomica della canna da zucchero autopoliploide Saccharum spontaneum", Nature Genetics 2022 54:6, 54(6), pp. 885–896. doi: 10.1038/s41588-022-01084-1.





