

TRASCRITOMICA
natura
COMUNICAZIONI
La caratterizzazione della trascrizione integrale della mutazione SF3B1 nella leucemia linfocitica cronica rivela una sottoregolazione degli introni trattenuti
Trascrizioni integrali| Sequenziamento dei nanopori| Analisi delle isoforme alternative
Sfondo
SÈ stato ampiamente riportato che le mutazioni matiche nel fattore di splicing SF3B1 si associano a vari tumori, tra cui la leucemia linfocitica cronica (LLC), il melanoma uveale, il cancro al seno, ecc. Inoltre, studi di trascrittomica a lettura breve hanno rivelato modelli di splicing aberranti indotti dalle mutazioni di SF3B1. Tuttavia, gli studi su questi modelli di splicing alternativo sono stati a lungo limitati al livello di evento e alla mancanza di conoscenza a livello di isoforma a causa della limitazione delle trascrizioni assemblate di breve lettura. Qui, è stata introdotta la piattaforma di sequenziamento dei nanopori per generare trascrizioni a lunghezza intera, che hanno consentito l'indagine sulle isoforme AS.
Progettazione sperimentale
Esperimenti
Raggruppamento:1. CLL-SF3B1(WT) 2. CLL-SF3B1(mutazione K700E); 3. Cellule B normali
Strategia di sequenziamento:Sequenziamento delle librerie 2D MinION, sequenziamento delle librerie 1D PromethION; dati di lettura breve dagli stessi campioni
Piattaforma di sequenziamento:ONT MiniONE; ONT Prometeo;
Analisi Bioinformatica
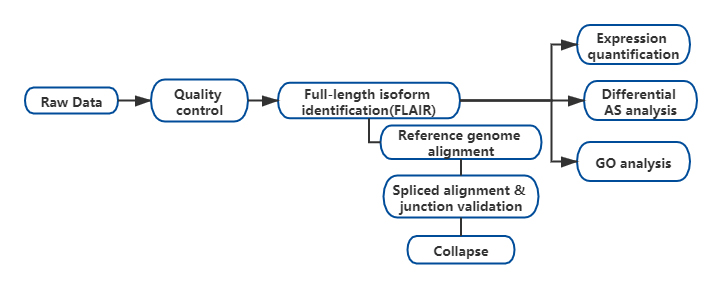
Risultati
UNsono state generate un totale di 257 milioni di letture da 6 campioni di CLL e 3 cellule B. In media il 30,5% di queste letture sono state identificate come trascrizioni integrali.
FL'analisi delle isoforme alternative a tutta lunghezza dell'RNA (FLAIR) è stata sviluppata per generare una serie di isoforme ad alta affidabilità. FLAIR può essere riassunto come:
Nallineamento delle letture di anopori: identificare la struttura generale della trascrizione basata sul genoma di riferimento;
Scorrezione della giunzione di giunzione: correggere gli errori di sequenza (rosso) con il sito di giunzione da introni annotati, introni da dati di lettura breve o entrambi;
Collasso: riassumere isoforme rappresentative basate su catene di giunzioni di giunzione (set di primo passaggio). Seleziona isofrom ad alta affidabilità in base al numero di letture supportate (soglia: 3).
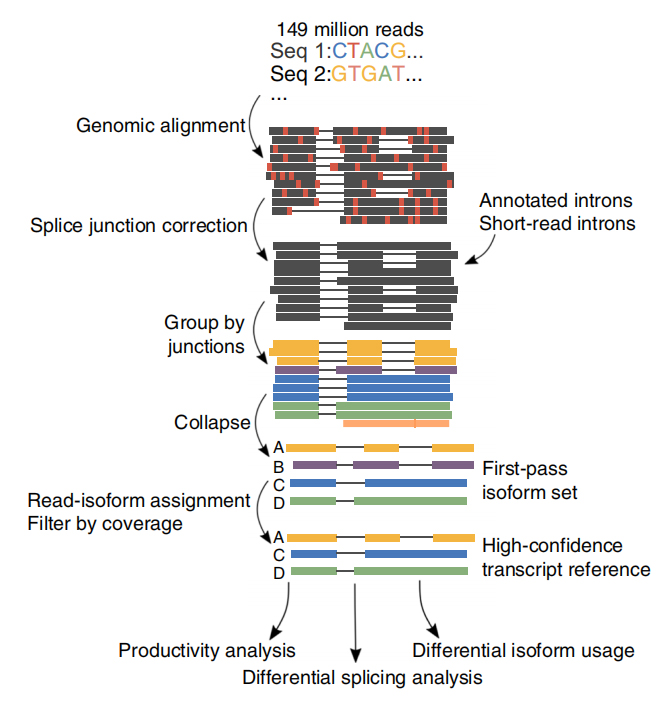
Figura 1. Analisi FLAIR per identificare le isoforme a lunghezza intera associate alla mutazione SF3B1 nella CLL
FLAIR ha identificato 326.699 isoforme di giunzione ad alta sicurezza, il 90% delle quali sono nuove isoforme. Si è scoperto che la maggior parte di queste isoforme non annotate erano nuove combinazioni di giunzioni di giunzione note (142.971), mentre le restanti nuove isoforme contenevano introne trattenuto (21.700) o nuovo esone (3594).
Lle sequenze a lunga lettura consentono l'identificazione dei siti di giunzione alterati da SF3B1-K700E mutanti a livello di isoforma. È stato riscontrato che 35 3'SS alternativi e 10 5'SS alternativi avevano uno splicing differenziale significativo tra SF3B1-K700E e SF3B1-WT. 33 delle 35 alterazioni sono state scoperte di recente da sequenze di lettura prolungata. Nei dati di Nanopore, la distribuzione della distanza tra i 3'SS alterati da SF3B1-K700E e i picchi dei siti canonici è di circa -20 bp, che è significativamente diversa da una distribuzione di controllo, simile a quanto riportato nelle sequenze di lettura breve della CLL. Sono state analizzate le isoforme del gene ERGIC3, dove una nuova isoforma contenente il sito di giunzione prossimale è stata trovata più abbondante in SF3B1-K700E. Sia i 3'SS prossimali che quelli distali erano associati a modelli AS distinti che generavano più isoforme.
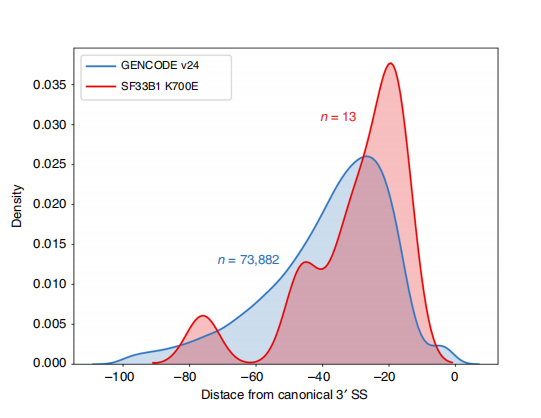
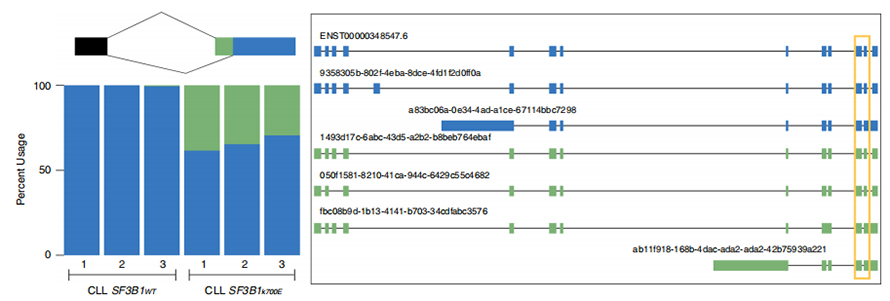
Figura 2. Modelli di splicing alternativi 3′ identificati con i dati di sequenziamento dei nanopori
L'analisi dell'utilizzo degli eventi IR è stata a lungo limitata nell'analisi basata su letture brevi a causa della fiducia nell'identificazione e quantificazione degli IR. L'espressione delle isoforme IR in SF3B1-K700E e SF3B1-WT è stata quantificata sulla base di sequenze di nanopori, rivelando una down-regulation globale delle isoforme IR in SF3B1-K700E.
Figura 4. Intensità agricola e connettività di rete in tre sistemi agricoli (A e B); Analisi casuale delle foreste (C) e relazione tra intensità agricola e colonizzazione AMF (D)
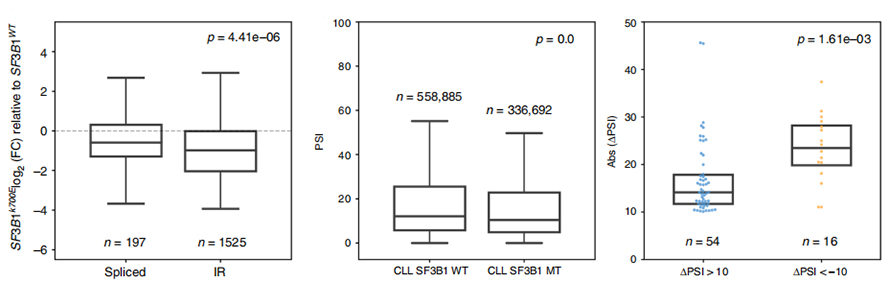
Figura 3. Gli eventi di ritenzione degli introni sono più fortemente sottoregolati nella CLL SF3B1-K700E
Tecnologia
Sequenziamento a lunga lettura di nanopori
NIl sequenziamento di anopori è una tecnologia di sequenziamento del segnale elettrico in tempo reale di una singola molecola.
DIl DNA o l'RNA a doppio filamento si legheranno alla proteina nanoporosa incorporata nel biofilm e si svolgeranno sotto la guida della proteina motrice.
DI filamenti di NA/RNA passano attraverso la proteina del canale dei nanopori a una certa velocità sotto l'azione della differenza di voltaggio.
Mle olecole generano segnali elettrici diversi a seconda della struttura chimica.
RIl rilevamento in tempo reale delle sequenze viene ottenuto tramite l'identificazione delle basi.
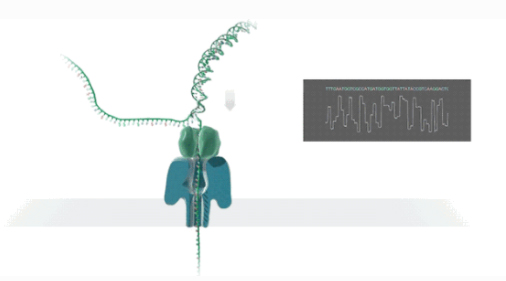
Esecuzione del sequenziamento del trascrittoma a lunghezza intera
√ Saturazione dei dati
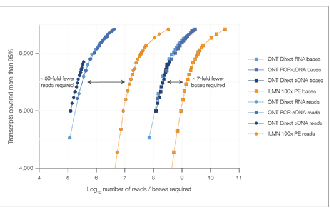
Sono necessarie 7 volte meno letture per raggiungere una saturazione dei dati paragonabile.
√ Identificazione della struttura della trascrizione
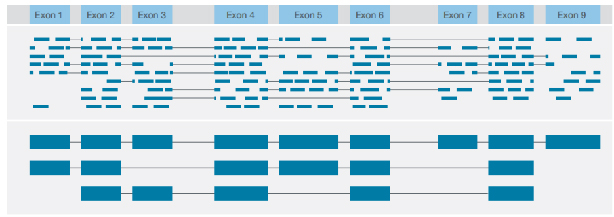
Identificazione di diverse varianti strutturali con lettura consensuale dell'intera lunghezza di ciascuna trascrizione
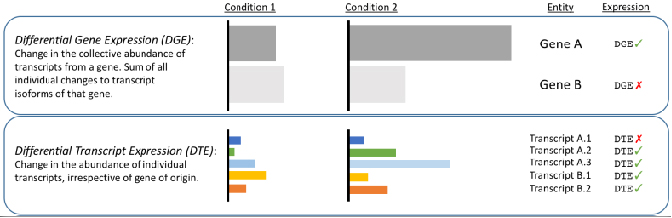
√ Analisi differenziale a livello di trascrizione: rivela i cambiamenti nascosti dalle letture brevi
Riferimento
Tang AD, Soulette CM, Baren MJV, e al. La caratterizzazione della trascrizione completa della mutazione SF3B1 nella leucemia linfocitica cronica rivela una sottoregolazione degli introni trattenuti[J]. Comunicazioni sulla natura.
Tecnologia e punti salienti mira a condividere le più recenti applicazioni di successo di diverse tecnologie di sequenziamento ad alto rendimento in vari ambiti di ricerca, nonché idee brillanti nella progettazione sperimentale e nel data mining.
Orario di pubblicazione: 08 gennaio 2022