

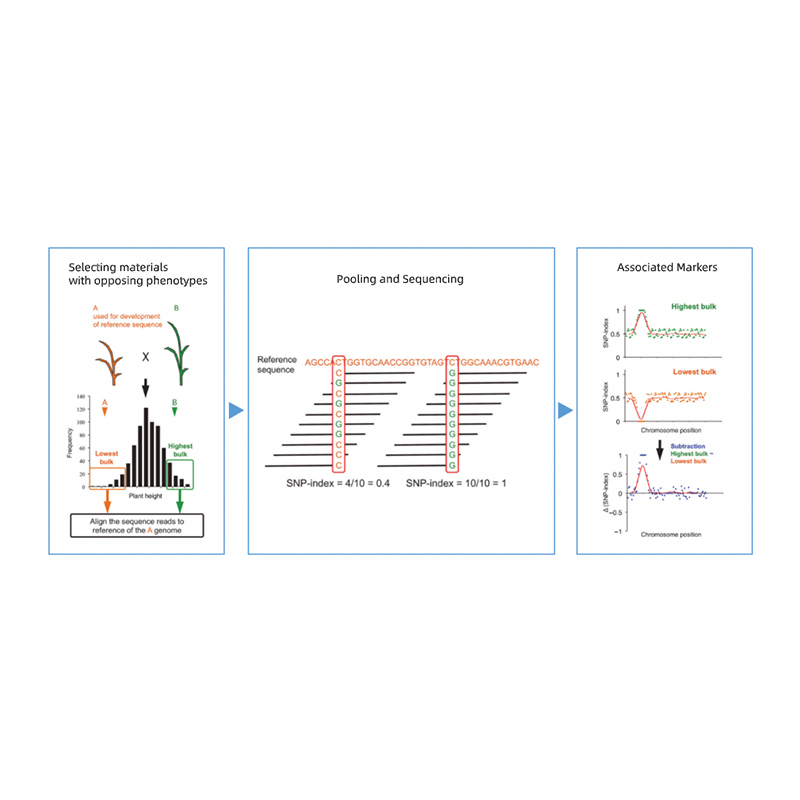
Analisi segregante massaggiata

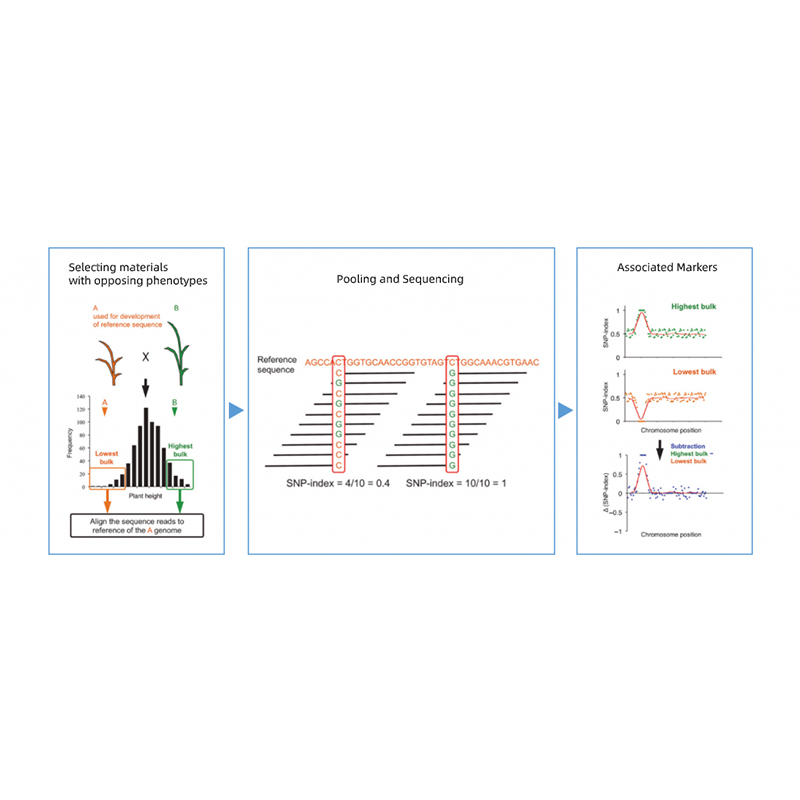
Vantaggi del servizio
Takagi et al., The Plant Journal, 2013
● Localizzazione accurata: miscelazione di massa con 30+30-200+200 persone per ridurre al minimo il rumore di fondo; Previsione della regione candidata non sinonimo di mutatanti.
● Analisi completa: annotazione della funzione genica candidata approfondita, tra cui NR, SwissProt, Go, Kegg, COG, KOG, ecc.
● Tempo di consegna più rapido: localizzazione del gene rapido entro 45 giorni lavorativi.
● Voga esperienza: BMK ha contribuito in migliaia di tratti di localizzazione, coprendo diverse specie come colture, prodotti acquatici, foreste, fiori, frutta, ecc.
Specifiche del servizio
Popolazione:
Separare la progenie dei genitori con fenotipi opposti.
EG F2 Progeny, Backcrossing (BC), linea inbred ricombinante (RIL)
Miscelazione della piscina
Per i tratti qualitativi: da 30 a 50 individui (minimo 20)/massa
Per Tratis quantitativi: da 5% al 10% individui con fenotipi estremi nell'intera popolazione (minimo 30+30).
Profondità di sequenziamento consigliato
Almeno 20x/genitore e 1x/prole individuale (ad es. Polle di miscelazione della prole di 30+30 individuo, la profondità di sequenziamento sarà 30x per massa)
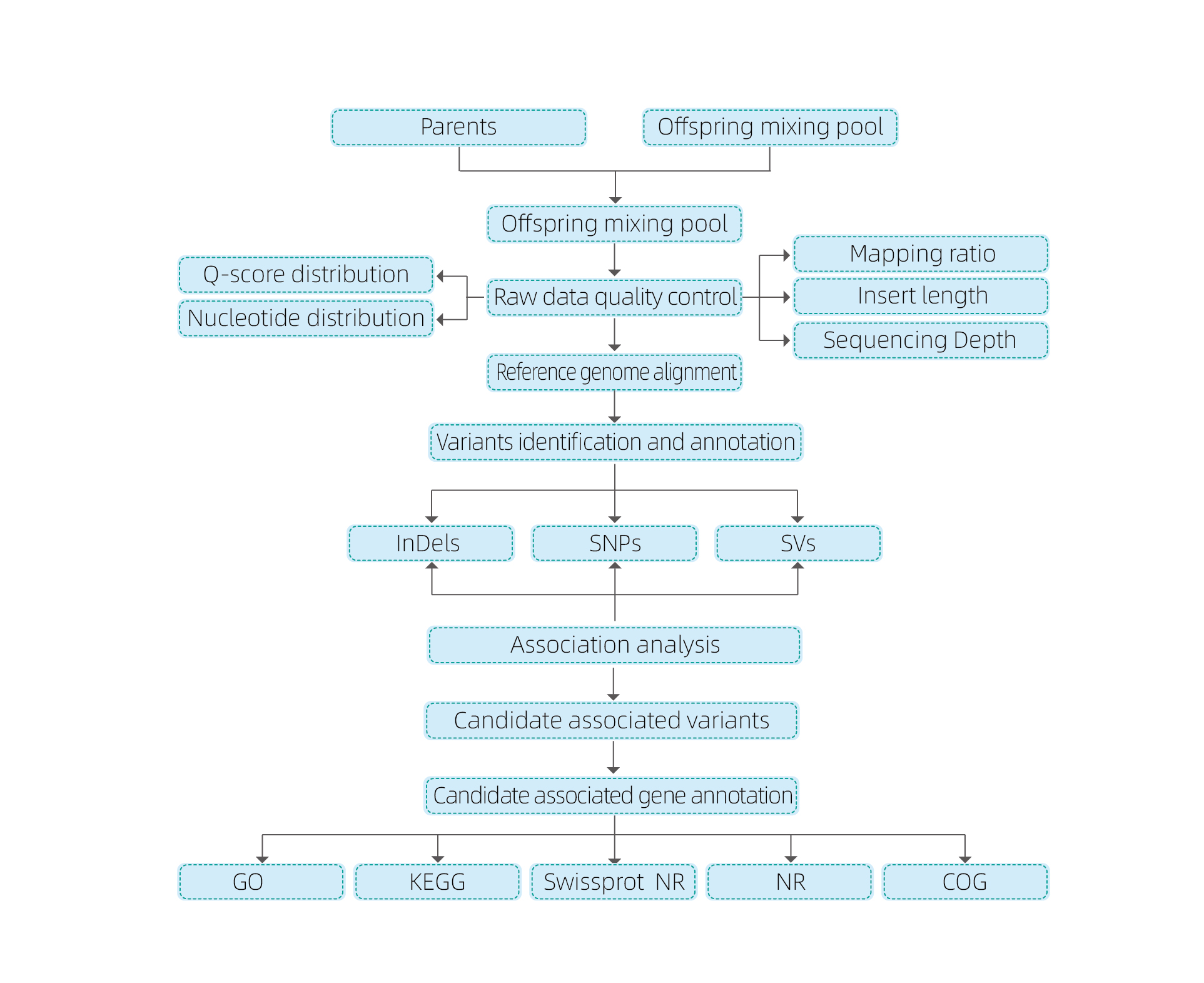
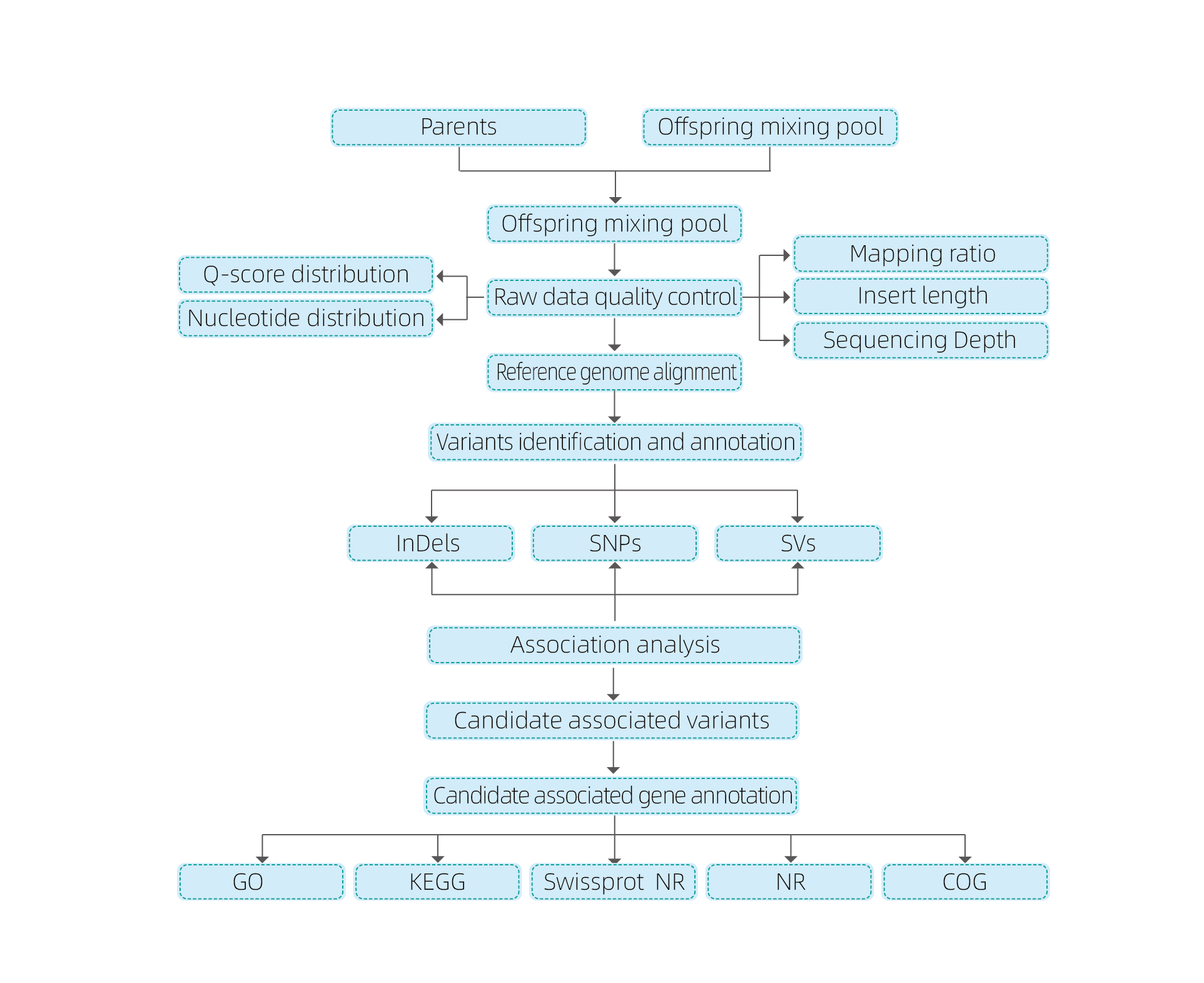
Analisi bioinformatiche
● Resequenziamento dell'intero genoma
● Elaborazione dei dati
● Chiamata SNP/Indel
● Screening della regione candidata
● Annotazione della funzione genica candidata
Requisiti di esempio e consegna
Requisiti del campione:
Nucleotidi:
| campione gDNA | Campione di tessuto |
| Concentrazione: ≥30 ng/μl | Piante: 1-2 g |
| Importo: ≥2 μg (Volumn ≥15 μL) | Animali: 0,5-1 g |
| PURITÀ: OD260/280 = 1.6-2.5 | Sangue intero: 1,5 ml |
Flusso di lavoro di servizio

Design dell'esperimento

Consegna del campione

Estrazione di RNA

Costruzione della biblioteca

Sequenziamento

Analisi dei dati

Servizi post-vendita
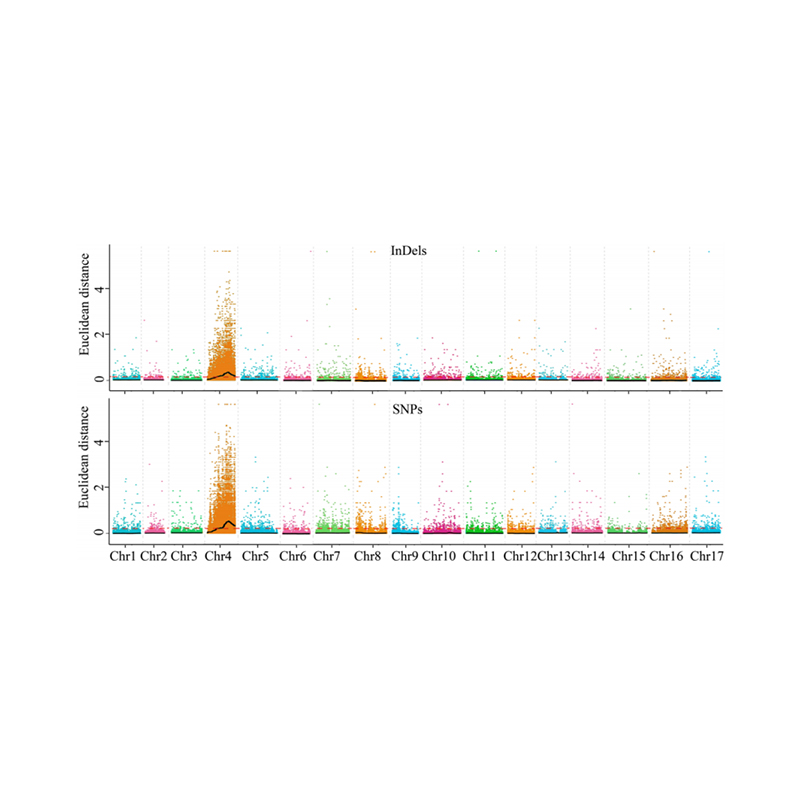
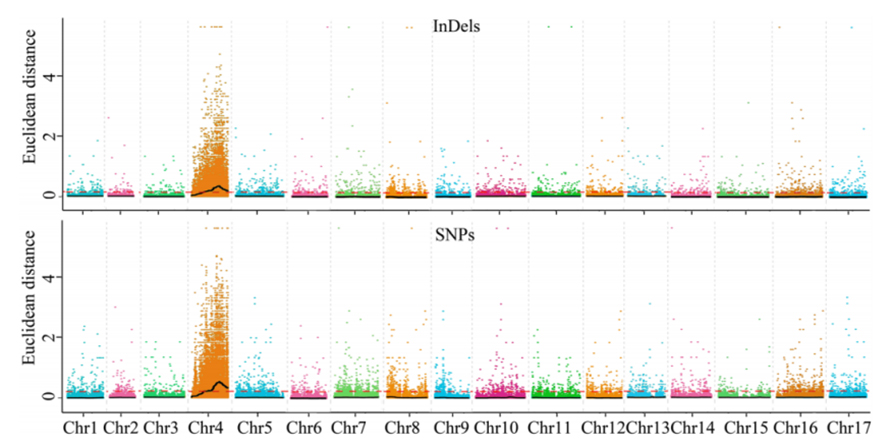
1. Base di analisi dell'associazione sulla distanza euclidea (DE) per identificare la regione candidata. Nella figura seguente
Asse X: numero cromosomico; Ogni punto rappresenta un valore ED di un SNP. La linea nera corrisponde al valore ED montato. Un valore ED più elevato indica un'associazione più significativa tra il sito e il fenotipo. La linea del cruscotto rosso rappresenta la soglia di un'associazione significativa.
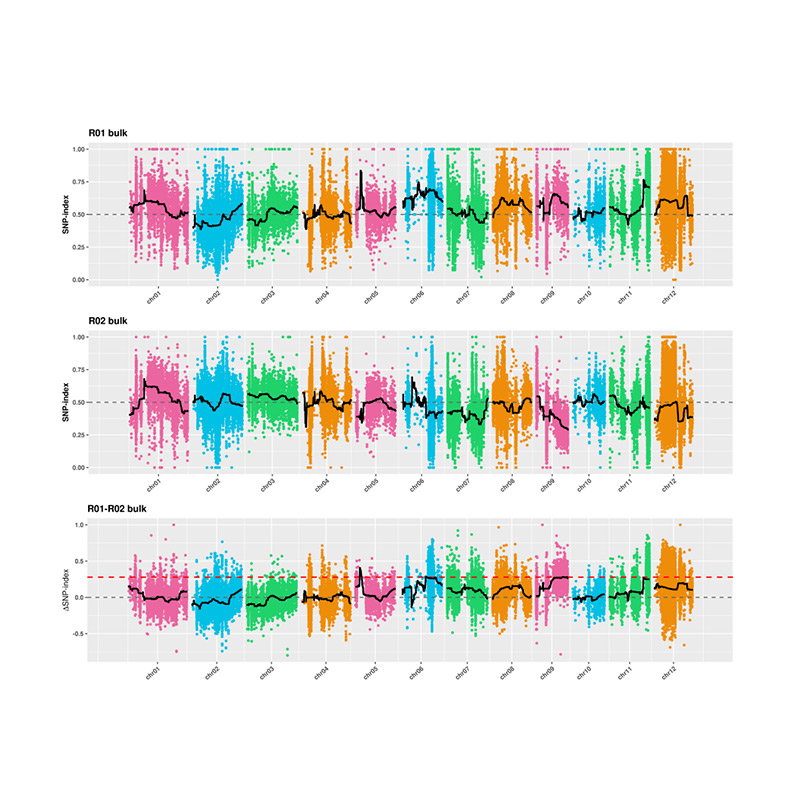
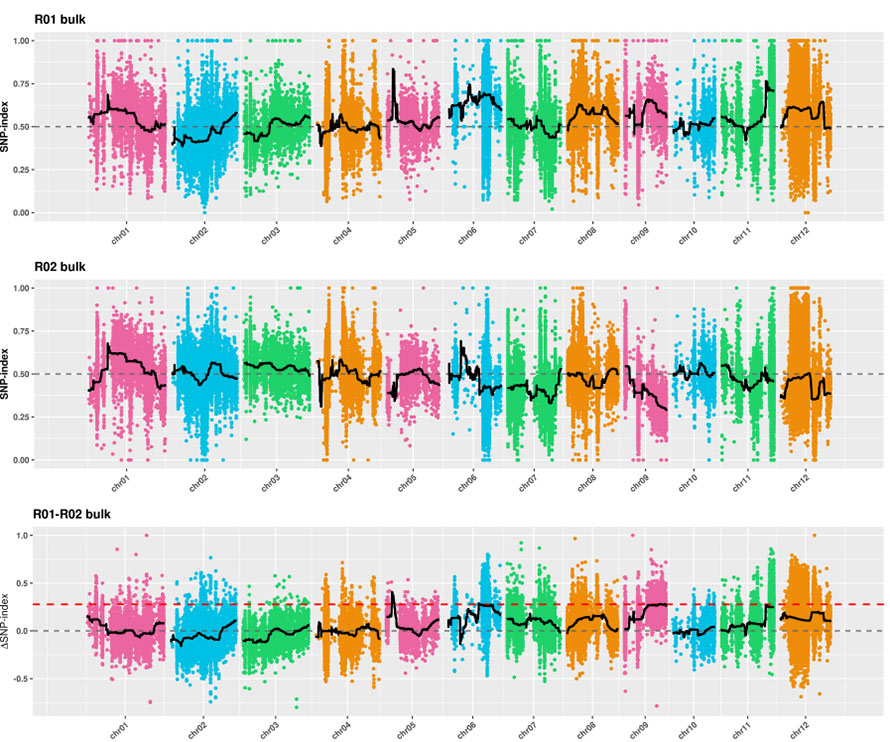
2. Analisi di associazione basata su nessuna indice SNP
Asse X: numero cromosomico; Ogni punto rappresenta il valore di indice SNP. La linea nera sta per il valore SNP-indice montato. Maggiore è il valore, più significativo è l'associazione.
Caso BMK
Il locus di tratto quantitativo dell'effetto maggiore fnl7.1 codifica una proteina abbondante di embriogenesi tardiva associata alla lunghezza del collo del frutto nel cetriolo
Pubblicato: Giornale di biotecnologia delle piante, 2020
Strategia di sequenziamento:
Genitori (Jin5-508, YN): tutto il genoma di resequenziamento per 34 × e 20 ×.
Pool di DNA (50 colpi lunghi e 50 a collo corto): resequenziamento per 61 × e 52 ×
Risultati chiave
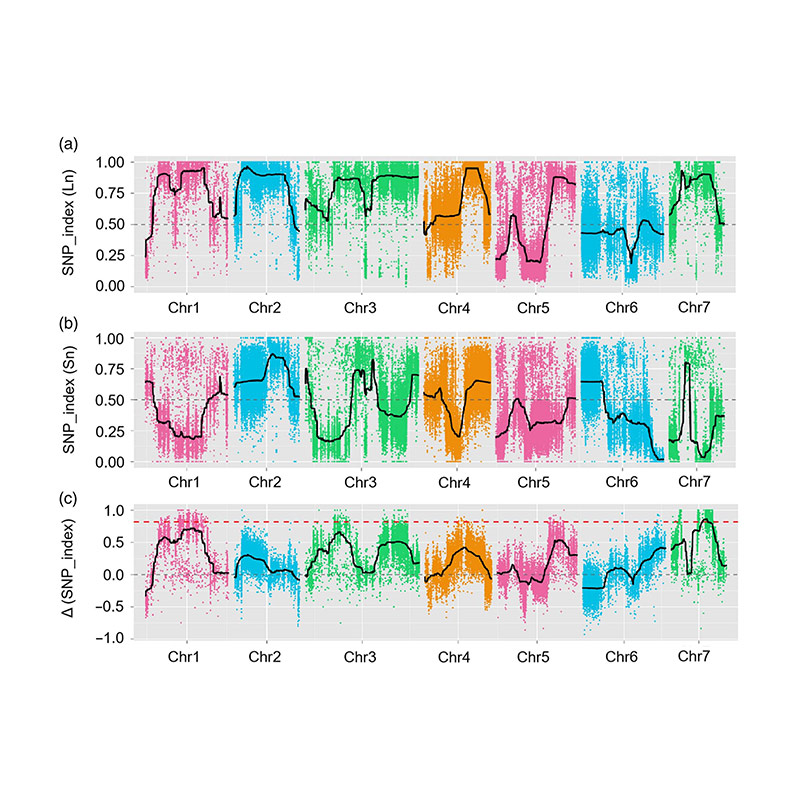
In questo studio, la popolazione segregata (F2 e F2: 3) è stata generata attraversando la linea di cetriolo a collo lungo Jin5-508 e YN a collo corto. Due pool di DNA sono state costruite da 50 individui estremi a collo lungo e 50 individui a collo corto. Il QTL a effetto maggiore è stato identificato su CHR07 mediante analisi BSA e mappatura QTL tradizionale. La regione candidata è stata ulteriormente ridotta da mappatura fine, quantificazione dell'espressione genica ed esperimenti transgenici, che hanno rivelato il gene chiave nel controllo della lunghezza del collo, CSFNL7.1. Inoltre, il polimorfismo nella regione del promotore CSFNL7.1 è risultato associato all'espressione corrispondente. Ulteriori analisi filogenetiche hanno suggerito che è molto probabile che il locus FNL7.1 sia originato dall'India.
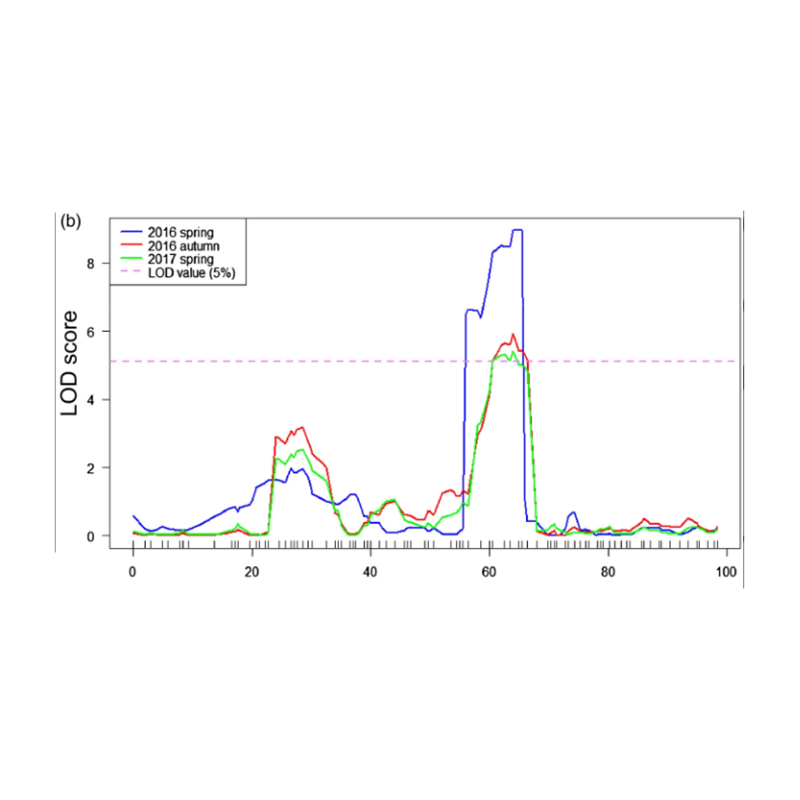
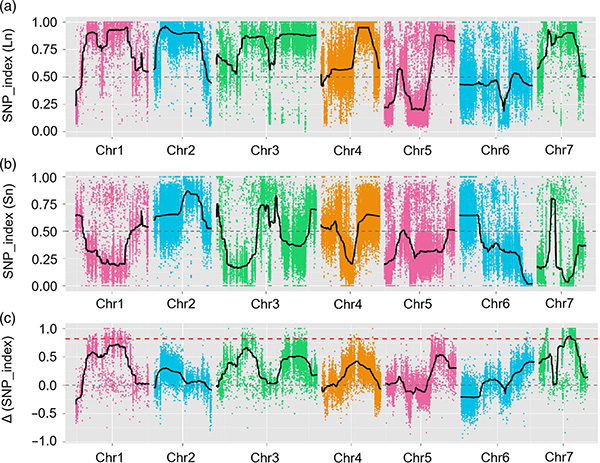 Mappatura QTL nell'analisi BSA per identificare la regione candidata associata alla lunghezza del collo del cetriolo | 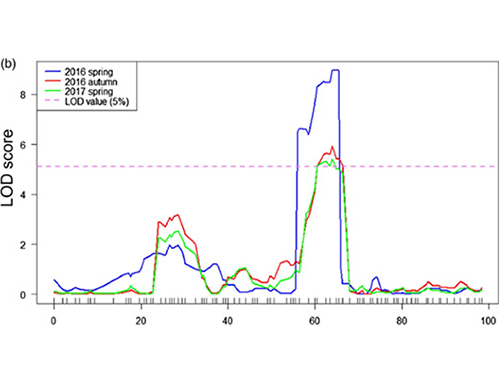 Profili LOD di QTL a lunghezza del collo di cetriolo identificati su Chr07 |
Xu, X., et al. "Il locus di tratto quantitativo dell'effetto maggiore FNL7.1 codifica una proteina abbondante di embriogenesi tardiva associata alla lunghezza del collo del frutto nel cetriolo." Plant Biotechnology Journal 18.7 (2020).



