10x Genomics Visio Visio Trascrittoma

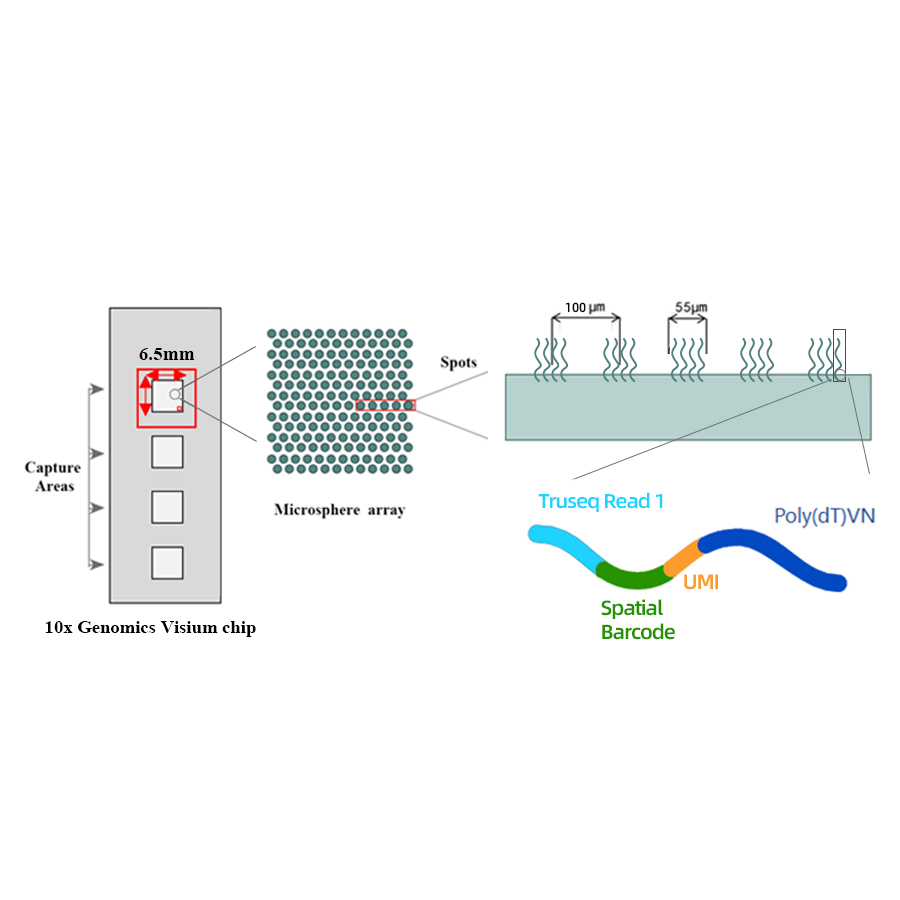
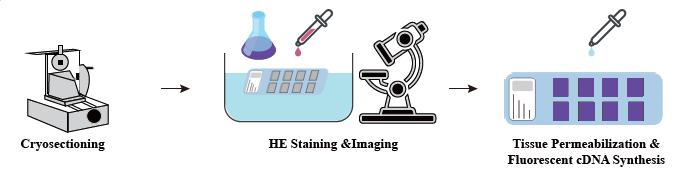
Schema tecnico

Caratteristiche
● Risoluzione: 100 µm
● Diametro spot: 55 µm
● Numero di punti: 4992
● Area di acquisizione: 6,5 x 6,5 mm
● Ogni punto con codice a barre è caricato con primer composti da 4 sezioni:
- coda poli (dt) per innesco di mRNA e sintesi di cDNA
- Identificatore molecolare unico (UMI) per correggere la distorsione dell'amplificazione
- Code spaziale
- Sequenza di legame del primer di sequenziamento di lettura 1 parziale
● Colorazione H&E delle sezioni
Vantaggi
●Servizio unico: integra tutte le fasi di esperienza e basate sulle abilità, tra cui criolezione, colorazione, ottimizzazione dei tessuti, codice a barre spaziale, preparazione della libreria, sequenziamento e bioinformatica.
● Team tecnico altamente qualificato: con esperienza in oltre 250 tipi di tessuti e oltre 100 specie tra cui umano, topo, mammifero, pesce e piante.
●Aggiornamento in tempo reale sull'intero progetto: con il pieno controllo del progresso sperimentale.
●Bioinformatica standard completa:Il pacchetto include 29 analisi e oltre 100 figure di alta qualità.
●Analisi e visualizzazione dei dati personalizzati: disponibile per diverse richieste di ricerca.
●Analisi articolare opzionale con sequenziamento mRNA a cellula singola
Specifiche
| Requisiti del campione | Biblioteca | Strategia di sequenziamento | Dati consigliati | Controllo di qualità |
| Campioni di crio incorporati da ottobre (Diametro ottimale: circa 6x6x6 mm³) 2 blocchi per campione | 10x Biblioteca di cDNA Visio | Illumina PE150 | 50k PE legge per punto (60 GB) | Rin> 7 |
Per maggiori dettagli sulla guida alla preparazione del campione e sul flusso di lavoro di servizio, non esitare a parlare con unEsperto di bmkgene
Flusso di lavoro di servizio
Nella fase di preparazione del campione, viene eseguita una sperimentazione iniziale di estrazione dell'RNA per garantire che sia possibile ottenere un RNA di alta qualità. Nella fase di ottimizzazione dei tessuti le sezioni sono colorate e visualizzate e le condizioni di permeabilizzazione per il rilascio di mRNA dal tessuto sono ottimizzate. Il protocollo ottimizzato viene quindi applicato durante la costruzione della libreria, seguito da sequenziamento e analisi dei dati.
Il flusso di lavoro completo del servizio prevede aggiornamenti in tempo reale e conferme dei clienti per mantenere un circuito di feedback reattivo, garantendo un'esecuzione regolare del progetto.


Include la seguente analisi:
Controllo della qualità dei dati:
o Output dei dati e distribuzione del punteggio di qualità
o Rilevamento genico per punto
o Copertura dei tessuti
Analisi del campione interno:
o Rich per la ricchezza genica
o Spot Clustering, inclusa l'analisi della dimensione ridotta
o Analisi dell'espressione differenziale tra cluster: identificazione dei geni marker
o Annotazione funzionale e arricchimento dei geni marcatori
Analisi tra gruppi
o Re-combinazione di punti da entrambi i campioni (ad es. Malatini e controlli) e RECLUSTER
o Identificazione dei geni marker per ciascun cluster
o Annotazione funzionale e arricchimento dei geni marcatori
o espressione differenziale dello stesso cluster tra i gruppi
Analisi del campione interno
Spot clustering
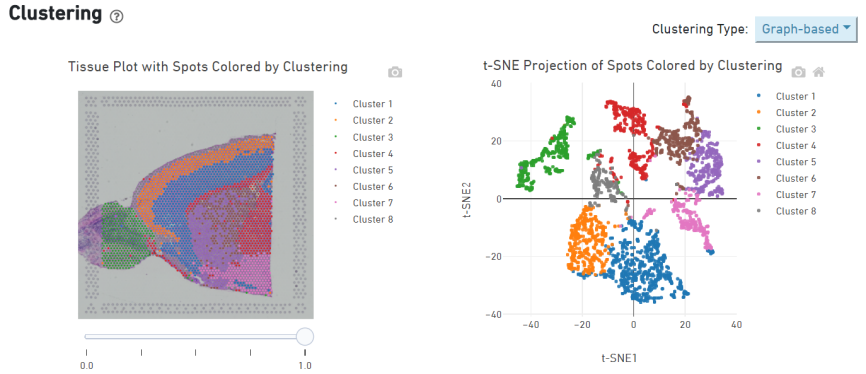
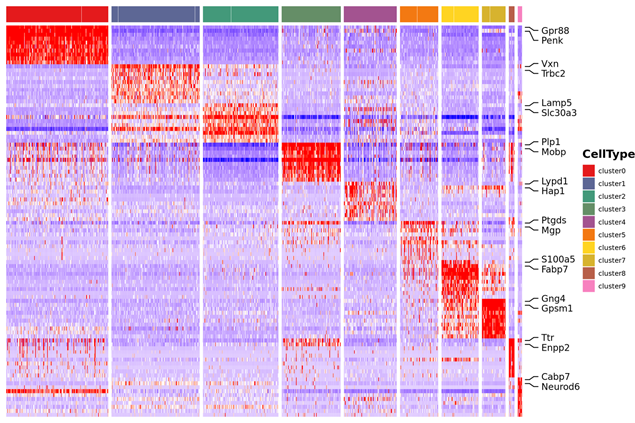
Identificazione dei geni marker e distribuzione spaziale

Analisi tra gruppi
Combinazione dei dati da entrambi i gruppi e re-cluster

Geni marker di nuovi cluster

Esplora i progressi facilitati dal servizio di trascrittomica spaziale di BMKGENE da 10x Visio in queste pubblicazioni in primo piano:
Chen, D. et al. (2023) 'MTHL1, un potenziale omologo Drosophila dei GPCR di adesione dei mammiferi, è coinvolto nelle reazioni antitumorali alle cellule oncogeniche iniettate nelle mosche',Atti della National Academy of Sciences degli Stati Uniti d'America, 120 (30), p. E2303462120. doi: /10.1073/pnas.2303462120
Chen, Y. et al. (2023) "L'acciaio consente la delineazione ad alta risoluzione di dati trascrittomici spazio-temporali",Briefing in bioinformatica, 24 (2), pagg. 1–10. doi: 10.1093/bavaglio/bbad068.
Liu, C. et al. (2022) "Un atlante spazio -temporale di organogenesi nello sviluppo di fiori di orchidee",Ricerca degli acidi nucleici, 50 (17), pagg. 9724-9737. doi: 10.1093/nar/gkac773.
Wang, J. et al. (2023) "L'integrazione della trascrittomica spaziale e del sequenziamento dell'RNA a nucleo singolo rivela le potenziali strategie terapeutiche per il leiomioma uterino",International Journal of Biological Sciences, 19 (8), pagg. 2515–2530. doi: 10.7150/ijbs.83510.