

Specific-Locus Amplified Fragment Sequencing (SLAF-Seq)

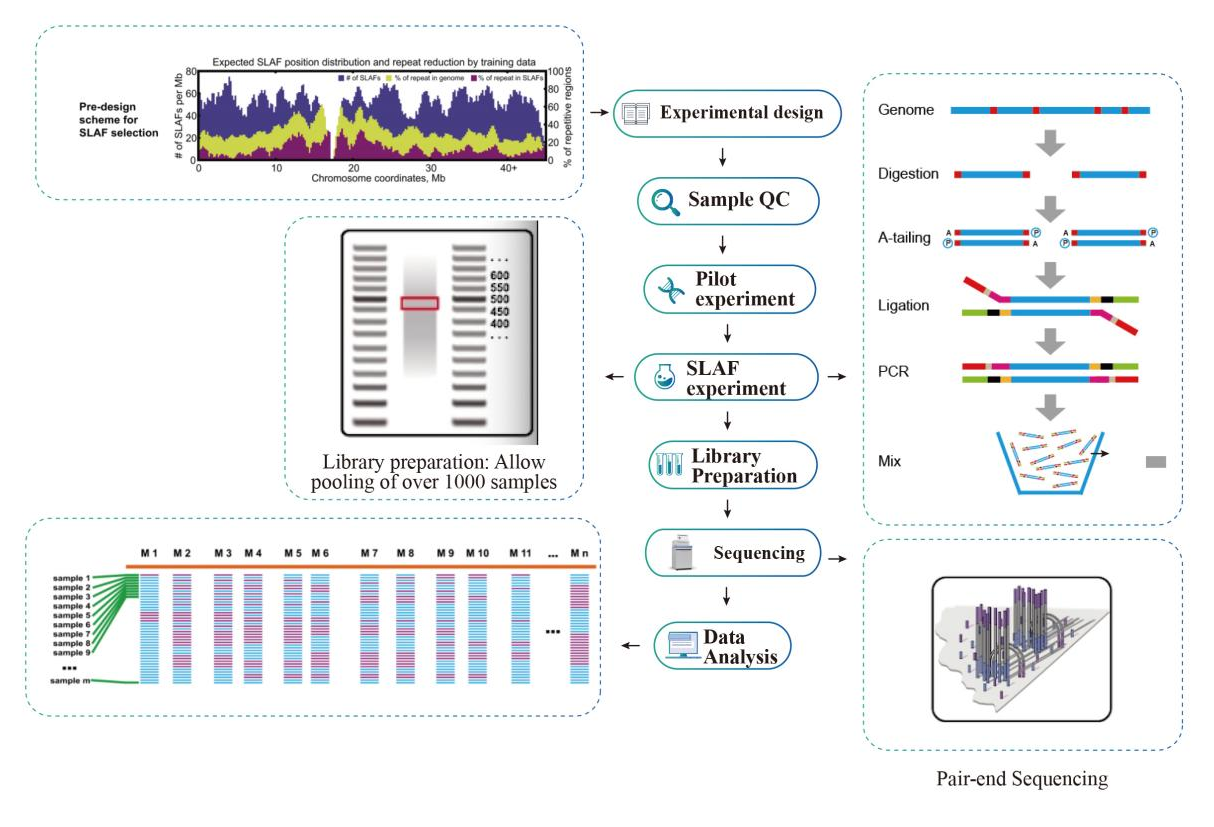
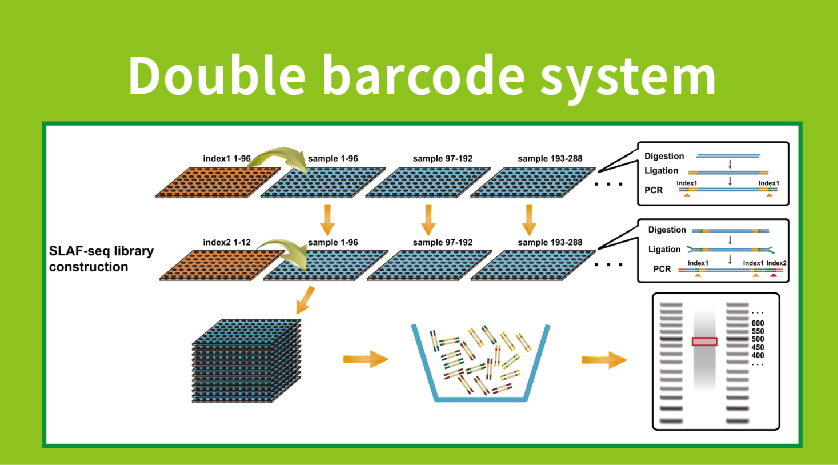
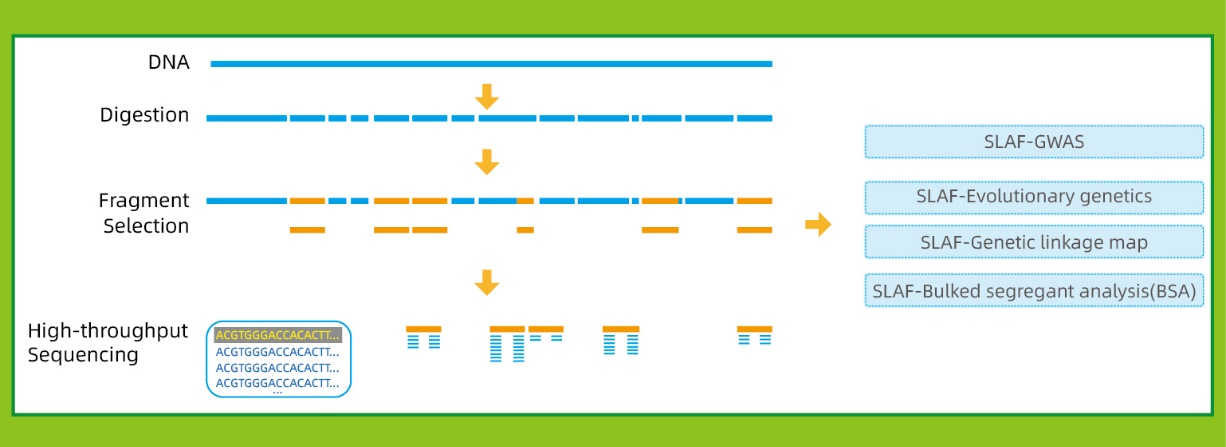
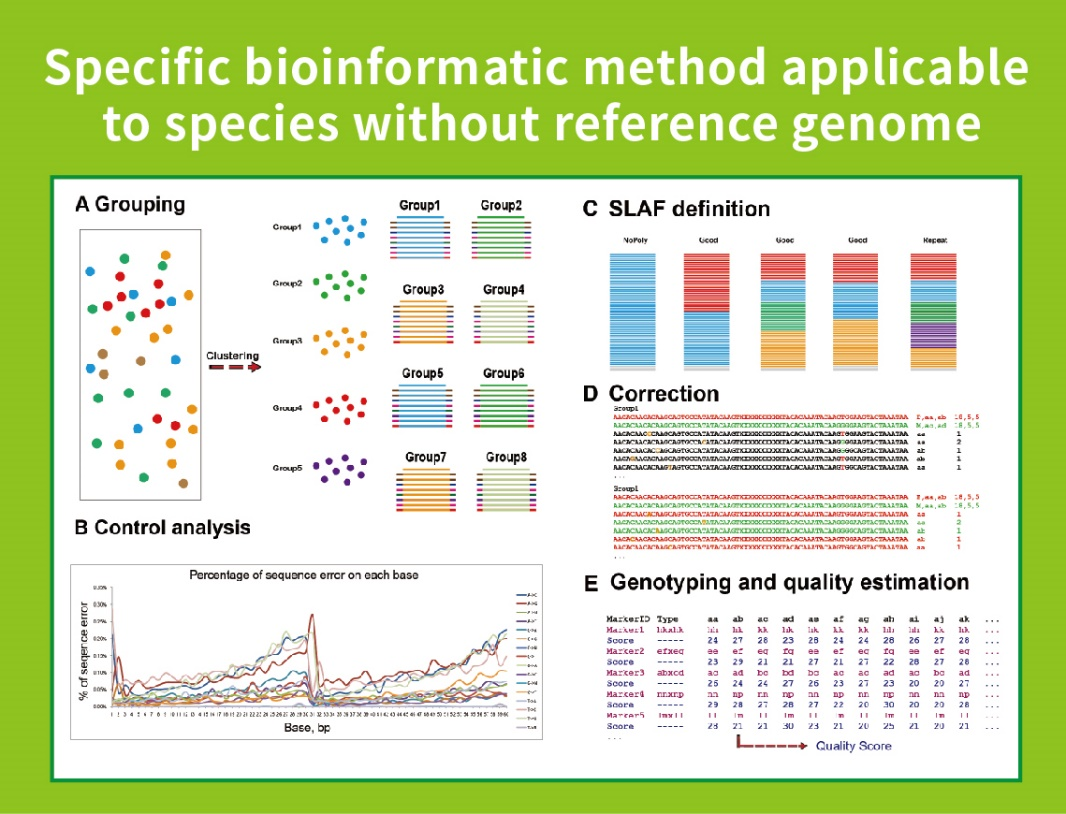
Munkafolyamat

Műszaki séma

Szolgáltatási jellemzők
● Szekvenálás a NovaSeq-en PE150-nel.
● Könyvtár előkészítés kettős vonalkóddal, amely több mint 1000 minta összegyűjtését teszi lehetővé.
● Ez a technika referencia genommal vagy anélkül is használható, esetenként eltérő bioinformatikai csővezetékekkel:
Referencia genommal: SNP és InDel felfedezés
Referenciagenom nélkül: minta klaszterezés és SNP felfedezés
● Ain silicoA tervezés előtti szakaszban a többszörös restrikciós enzimkombinációkat átvizsgálják, hogy megtalálják azokat, amelyek az SLAF címkék egyenletes eloszlását generálják a genom mentén.
● Az előkísérlet során három enzimkombinációt tesztelnek 3 mintában, így 9 SLAF-könyvtárat állítanak elő, és ezt az információt használják fel a projekt számára optimális restrikciós enzimkombináció kiválasztásához.
Szolgáltatás előnyei
●Magas genetikai marker felfedezése: A nagy áteresztőképességű kettős vonalkód-rendszer integrálása lehetővé teszi nagy populációk egyidejű szekvenálását, a lókusz-specifikus erősítés pedig növeli a hatékonyságot, biztosítva, hogy a címkeszámok megfeleljenek a különböző kutatási kérdések változatos követelményeinek.
● Alacsony genomfüggőség: Referenciagenommal rendelkező vagy anélküli fajokra alkalmazható.
●Rugalmas sématervezés: Egyenzimes, kettős enzimes, több enzimes emésztés és különféle típusú enzimek egyaránt kiválaszthatók, hogy megfeleljenek a különböző kutatási céloknak vagy fajoknak. Ain silicoelőtervezés történik az optimális enzimtervezés biztosítása érdekében.
● Nagy hatékonyság az enzimatikus emésztésben: Az an. vezetésein silicoAz előzetes tervezés és az előzetes kísérlet biztosította az optimális tervezést az SLAF-címkék egyenletes eloszlásával a kromoszómán (1 SLAF-címke/4Kb) és csökkentett ismétlődő szekvenciával (<5%).
●Széleskörű Szakértelem: Csapatunk rengeteg tapasztalatot hoz minden projektbe, több mint 5000 SLAF-Seq projektet zártak le több száz fajjal, beleértve a növényeket, emlősöket, madarakat, rovarokat és vízi szervezeteket.
● Saját fejlesztésű bioinformatikai munkafolyamat: A BMKGENE integrált bioinformatikai munkafolyamatot fejlesztett ki az SLAF-Seq számára, hogy biztosítsa a végső kimenet megbízhatóságát és pontosságát.
Szolgáltatási specifikációk
| Az elemzés típusa | Javasolt populációs skála | Szekvenálási stratégia | |
| A címkesorrend mélysége | Címkeszám | ||
| Genetikai térképek | 2 szülő és >150 utód | Szülők: 20x WGS Utódok: 10x | Genom mérete: <400 Mb: WGS ajánlott <1 Gb: 100 000 címke 1-2Gb:: 200K címke >2 Gb: 300 000 címke Max 500 ezer címke |
| Genome-Wide Association Studies (GWAS) | ≥200 minta | 10x | |
| Genetikai evolúció | ≥30 minta, minden alcsoportból >10 mintával | 10x | |
Szolgáltatási követelmények
Koncentráció ≥ 5 ng/µL
Teljes mennyiség ≥ 80 ng
Nanodrop OD260/280=1,6-2,5
Agaróz gél: nincs bomlás vagy szennyeződés, vagy csak korlátozott mértékben
Javasolt mintaszállítás
Tartály: 2 ml-es centrifugacső
(A legtöbb minta esetében azt javasoljuk, hogy ne tárolja etanolban)
Mintacímkézés: A mintákat egyértelműen fel kell címkézni, és meg kell egyeznie a benyújtott mintainformációs űrlappal.
Szállítás: Szárazjég: A mintákat először zsákokba kell csomagolni, és szárazjégbe kell temetni.
Szolgáltatási munkafolyamat






Minta minőségellenőrzés
Kísérleti kísérlet
SLAF-kísérlet
Könyvtár előkészítése
Sorrendezés
Adatelemzés
Értékesítés utáni szolgáltatások
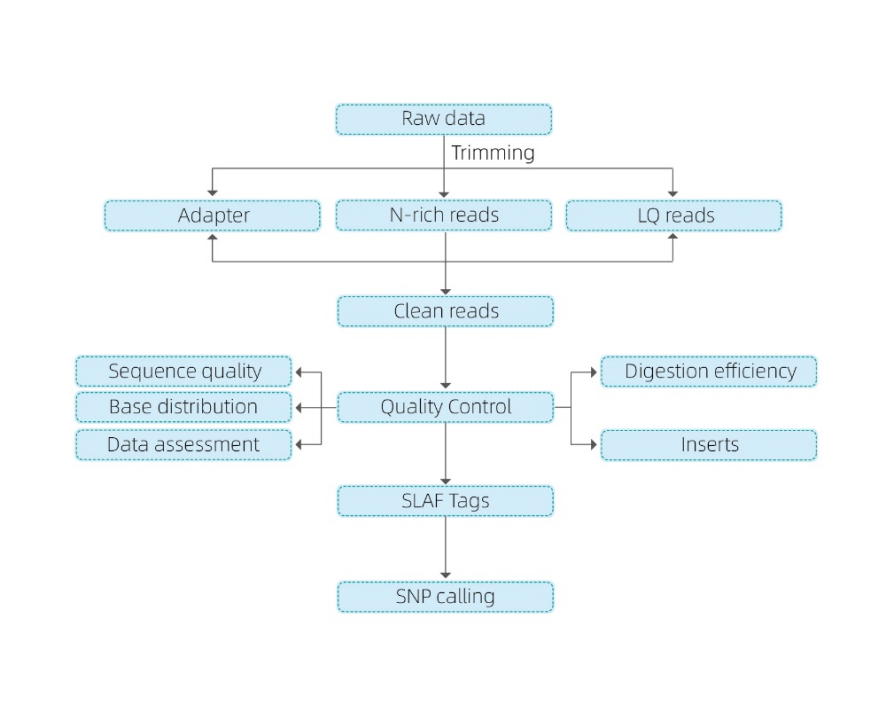 A következő elemzést tartalmazza:
A következő elemzést tartalmazza:

- Sorozati adatok minőségellenőrzése
- SLAF címke fejlesztés
Leképezés a referencia genomhoz
Referenciagenom nélkül: klaszterezés
- SLAF címkék elemzése.: statisztika, eloszlás a genomban
- Marker felfedezés: SNP, InDel, SNV, CV hívás és annotáció
Az SLAF címkék eloszlása a kromoszómákon:

Az SNP-k megoszlása a kromoszómákon:
 SNP annotáció
SNP annotáció

| Év | Folyóirat | IF | Cím | Alkalmazások |
| 2022 | Természeti kommunikáció | 17.694 | A bazsarózsa giga-kromoszómáinak és giga-genomjának genomi alapja Paeonia ostii | SLAF-GWAS |
| 2015 | Új fitológus | 7.433 | A háziasítási lábnyomok agronómiai jelentőségű genomiális régiókat rögzítenek szójabab | SLAF-GWAS |
| 2022 | Journal of Advanced Research | 12.822 | A Gossypium barbadense mesterséges introgressziói a G. hirsutumba az egész genomra kiváló lókuszokat tár fel a pamutszál minőségének és hozamának egyidejű javítása érdekében vonások | SLAF-Evolúciós genetika |
| 2019 | Molekuláris növény | 10.81 | A populációgenomikai elemzés és a De Novo Assembly feltárja Weedy eredetét A rizs, mint evolúciós játék | SLAF-Evolúciós genetika |
| 2019 | Természetgenetika | 31.616 | A ponty, a Cyprinus carpio genomszekvenciája és genetikai változatossága | SLAF-Linkage térkép |
| 2014 | Természetgenetika | 25.455 | A termesztett földimogyoró genomja betekintést nyújt a hüvelyes kariotípusokba, a poliploidokba evolúció és termény háziasítása. | SLAF-Linkage térkép |
| 2022 | Plant Biotechnology Journal | 9.803 | Az ST1 azonosítása feltár egy szelekciót, amely magában foglalja a vetőmag morfológiájának stoppolását és olajtartalom a szójabab háziasítása során | SLAF-Marker fejlesztés |
| 2022 | International Journal of Molecular Sciences | 6.208 | A Wheat-Leymus mollis 2Ns (2D) azonosítása és DNS-marker fejlesztése Diszómiás kromoszóma helyettesítés | SLAF-Marker fejlesztés |
| Év | Folyóirat | IF | Cím | Alkalmazások |
| 2023 | A növénytudomány határai | 6.735 | A cukortartalom QTL-térképezése és transzkriptom elemzése a Pyrus pyrifolia gyümölcsérése során | Genetikai térkép |
| 2022 | Plant Biotechnology Journal | 8.154 | Az ST1 azonosítása olyan szelekciót tár fel, amely magában foglalja a magok morfológiájának és olajtartalmának stoppolását a szójabab háziasítása során
| SNP hívás |
| 2022 | A növénytudomány határai | 6.623 | Genome-Wide Association Mapping of Hulless Barely Phenotypes in Drought Environment.
| GWAS |





