

Análise segregada

Vantaxes do servizo
Takagi et al., The Plant Journal, 2013
● Localización precisa: mesturar agrupacións con 30+30 a 200+200 individuos para minimizar o ruído de fondo; Predición da rexión candidata baseada en mutatantes non sinónimos.
● Análise completa: anotación de funcións xénicas candidatas en profundidade, incluíndo NR, SwissProt, GO, KEGG, COG, KOG, etc.
● Tempo de xiro máis rápido: localización de xenes rápida dentro de 45 días hábiles.
● Experiencia extensa: BMK contribuíu en miles de trazos de localización, abarcando especies diversas como cultivos, produtos acuáticos, bosque, flores, froitas, etc.
Especificacións de servizo
Poboación:
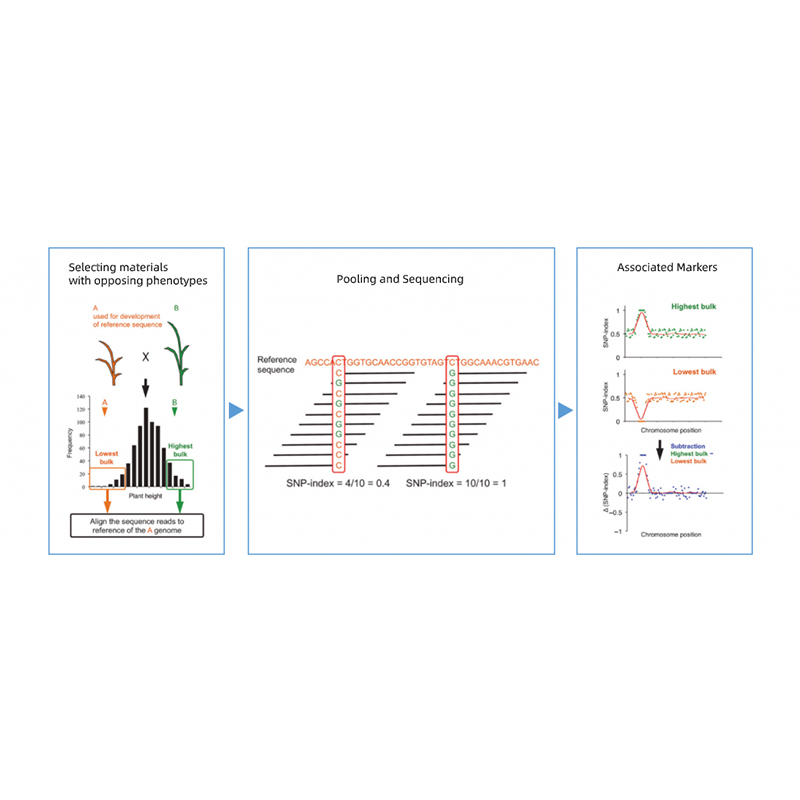
Segregando a descendencia de pais con fenotipos opostos.
Por exemplo, descendencia F2, retroceso (BC), liña de cría recombinante (RIL)
Piscina de mestura
Para trazos cualitativos: 30 a 50 individuos (mínimo 20)/a granel
Para Tratis cuantitativo: o 5% superior ao 10% dos individuos con fenotipos extremos en toda a poboación (mínimo 30+30).
Profundidade de secuenciación recomendada
Polo menos 20x/pai e 1x/descendencia individual (por exemplo, un grupo de mestura de descendencia de 30+30 individuais, a profundidade de secuenciación será de 30x por granel)
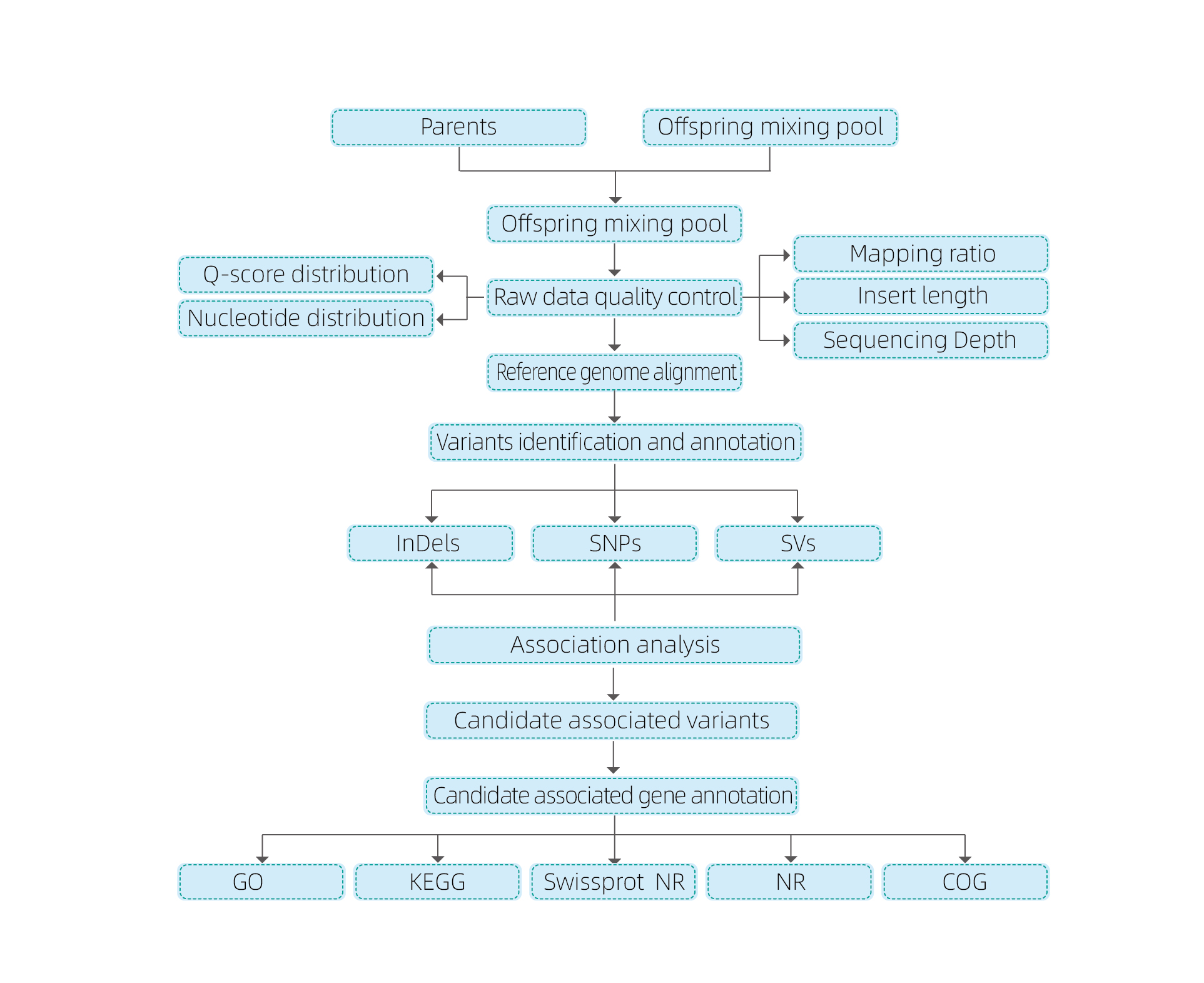
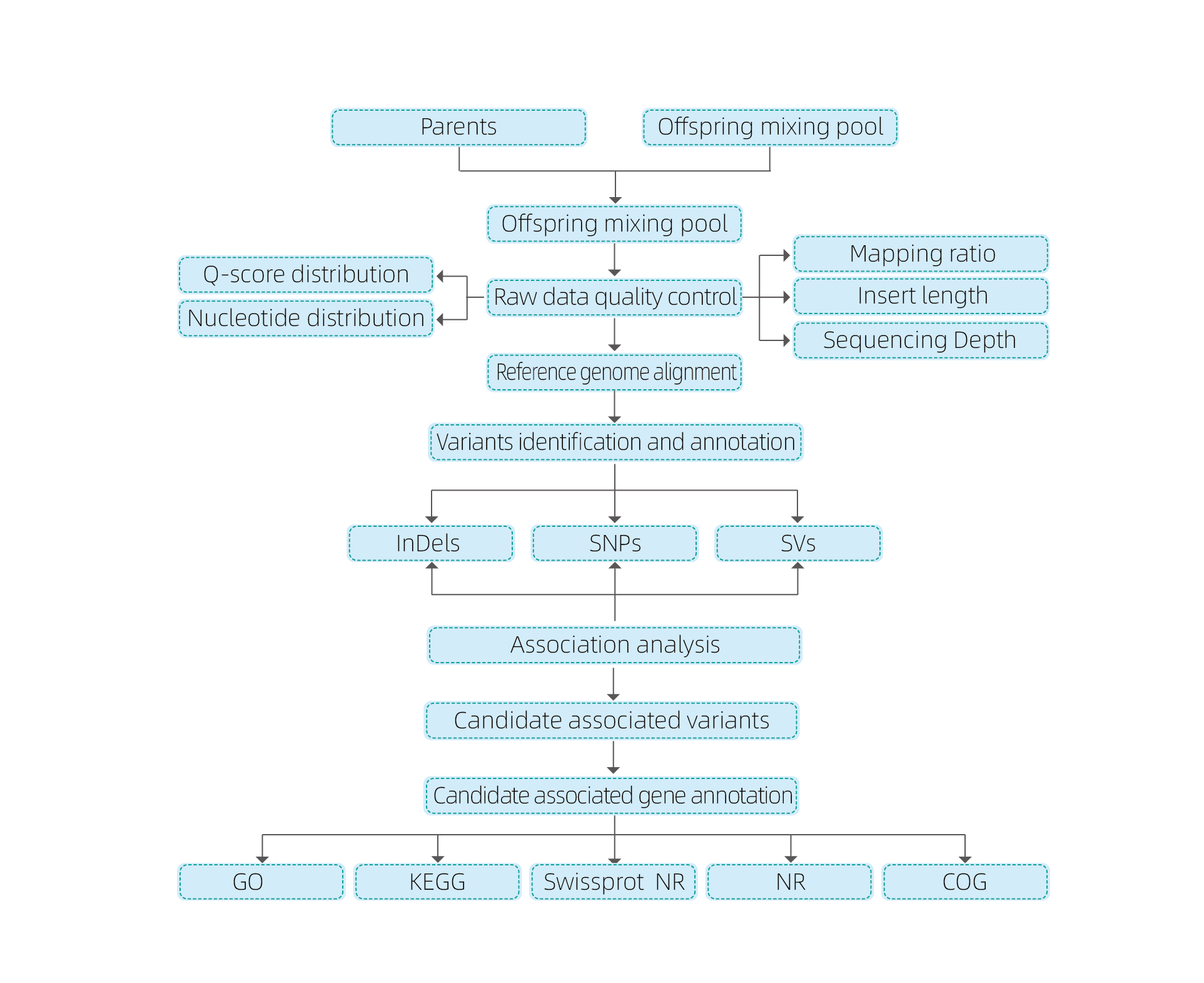
Análises de bioinformática
● Revisión do xenoma enteiro
● Procesamento de datos
● Chamadas SNP/Indel
● Screening da rexión candidata
● anotación da función xénica candidata
Requisitos de mostra e entrega
Requisitos da mostra:
Nucleótidos:
| Mostra de GDNA | Mostra de tecidos |
| Concentración: ≥30 ng/µl | Plantas: 1-2 g |
| Cantidade: ≥2 μg (volumn ≥15 µl) | Animais: 0,5-1 g |
| Pureza: OD260/280 = 1,6-2,5 | Sangue enteiro: 1,5 ml |
Fluxo de traballo de servizo

Deseño de experimentos

Entrega de mostra

Extracción de ARN

Construción da biblioteca

Secuenciación

Análise de datos

Servizos despois da venda
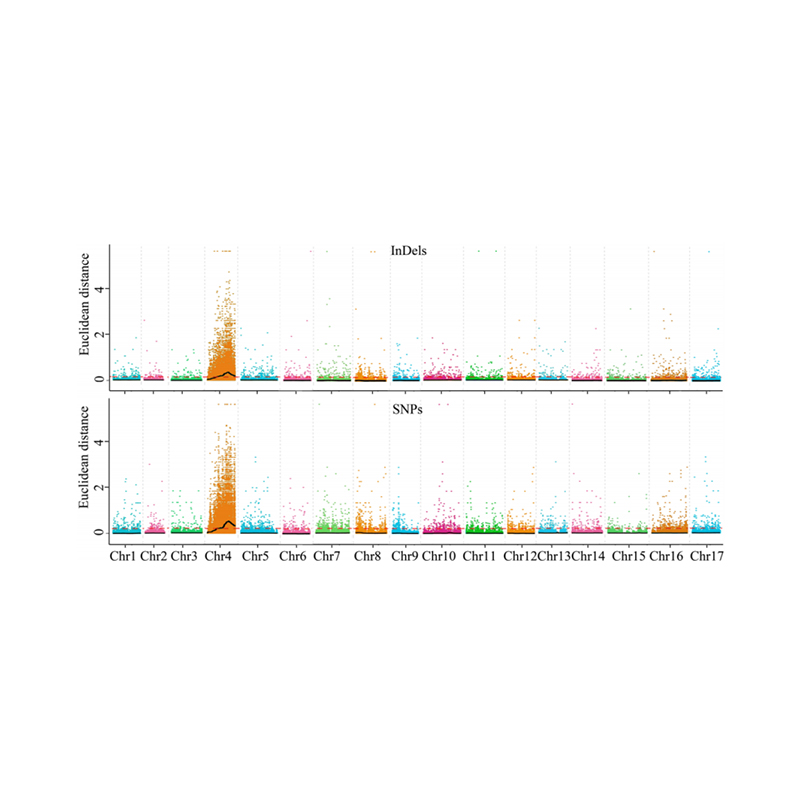
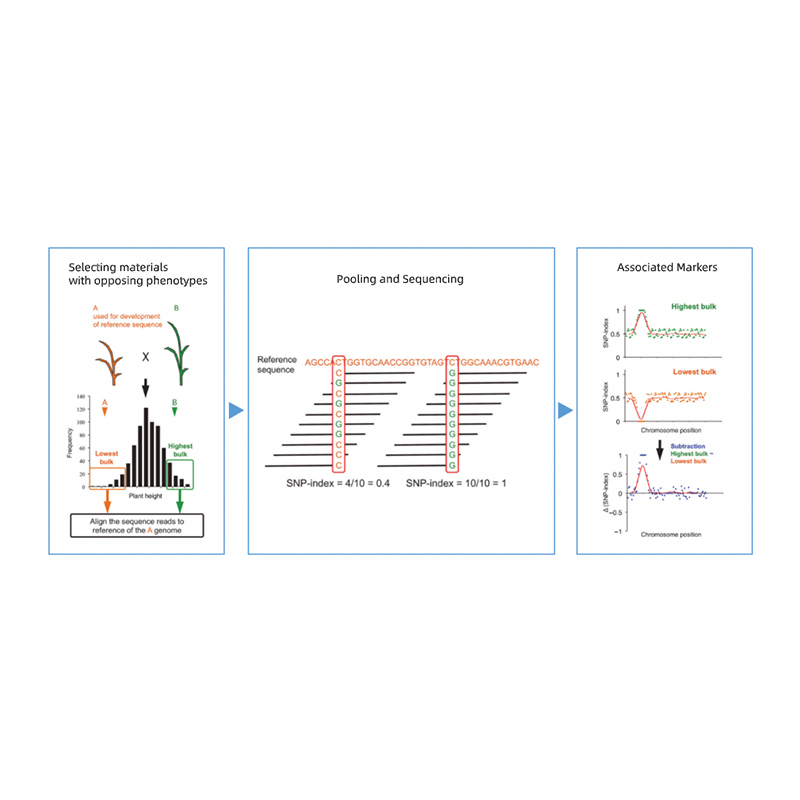
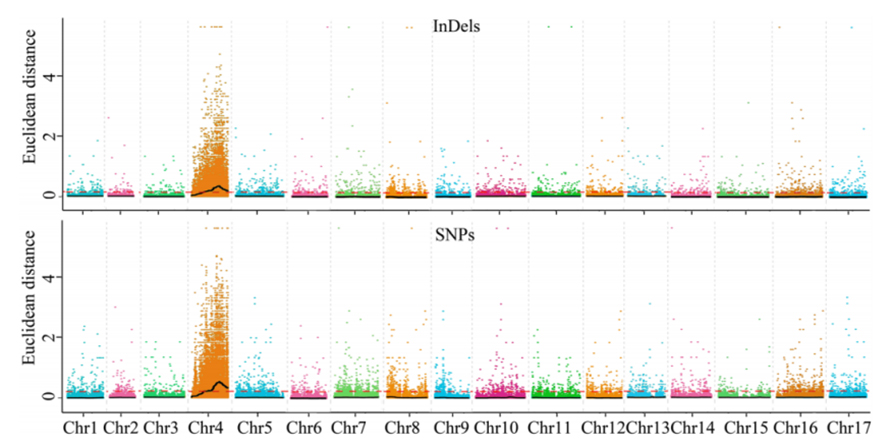
1. Base de análise de asociación na distancia euclidiana (ED) para identificar a rexión candidata. Na seguinte figura
X-eixe: número de cromosoma; Cada punto representa un valor ED dun SNP. A liña negra corresponde ao valor ED encaixado. Un maior valor ED indica unha asociación máis significativa entre o sitio e o fenotipo. A liña de guión vermello representa o limiar de asociación significativa.
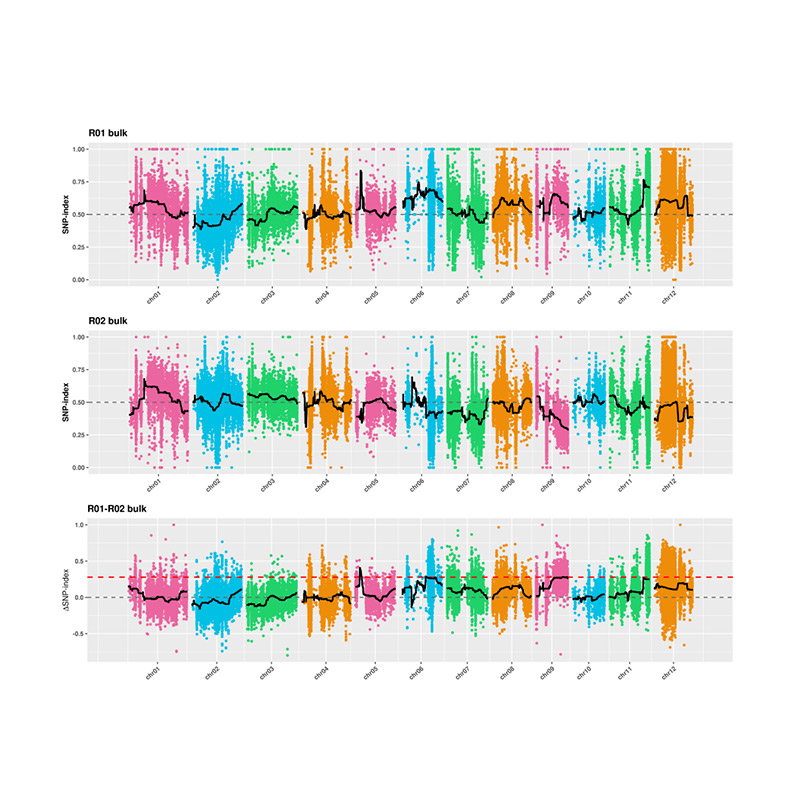
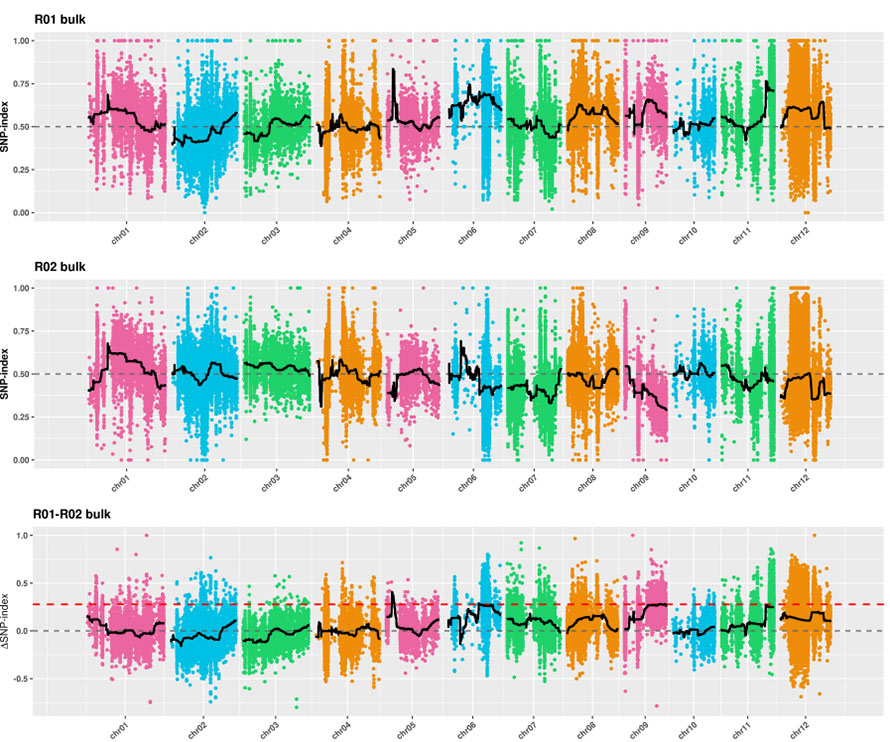
2. A análise de clasificación baseada no SNP-Index
X-eixe: número de cromosoma; Cada punto representa o valor do índice SNP. A liña negra significa o valor do índice SNP encaixado. Canto maior sexa o valor, máis significativa é a asociación.
Caso BMK
O trazo cuantitativo de maior efecto Locus FNL7.1 codifica unha proteína abundante da embrioxénese tardía asociada á lonxitude do pescozo do pepino
Publicado: Xornal de biotecnoloxía vexetal, 2020
Estratexia de secuenciación:
Pais (JIN5-508, YN): Genoma enteiro de recursos para 34 × e 20 ×.
Piscinas de ADN (50 pescozo longo e 50 pescozo curto): RESEQUENCIA PARA 61 × e 52 ×
Resultados clave
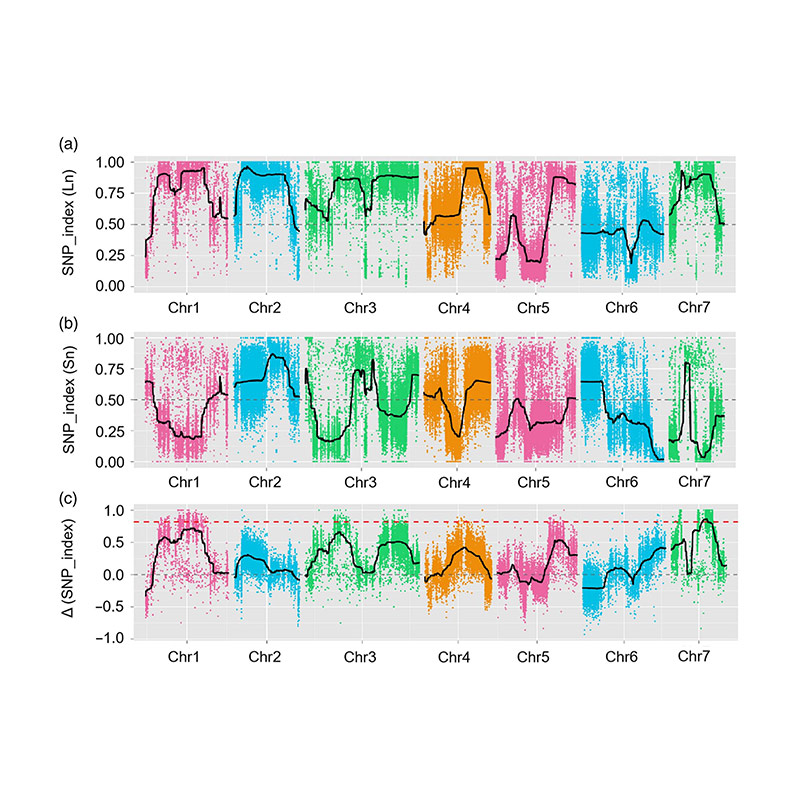
Neste estudo, a poboación segregación (F2 e F2: 3) foi xerada ao cruzar a liña de pepino de pescozo Jin5-508 e o pescozo curto Yn. Dúas piscinas de ADN foron construídas por 50 individuos extremos de pescozo e 50 individuos extremos de pescozo. O QTL de efecto principal foi identificado en CHR07 mediante a análise BSA e o mapeo tradicional de QTL. A rexión candidata foi reducida aínda máis mediante o mapeo fino, a cuantificación da expresión xénica e os experimentos transxénicos, que revelaron o xene clave no control da lonxitude do pescozo, CSFNL7.1. Ademais, o polimorfismo na rexión promotora CSFNL7.1 atopouse asociado á expresión correspondente. Unha maior análise filoxenética suxeriu que o lugar FNL7.1 é moi probable que se orixine na India.
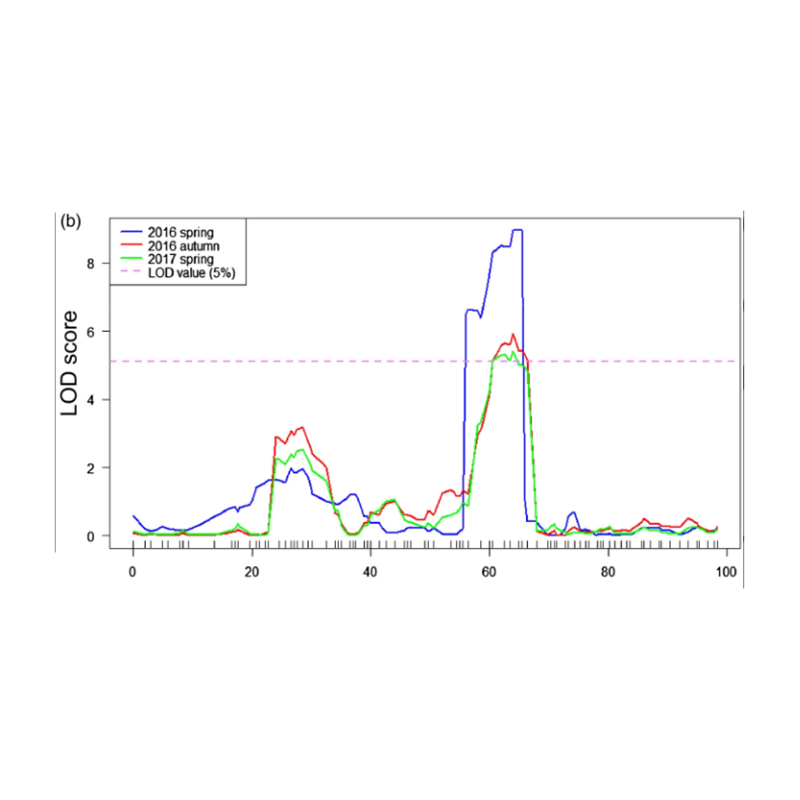
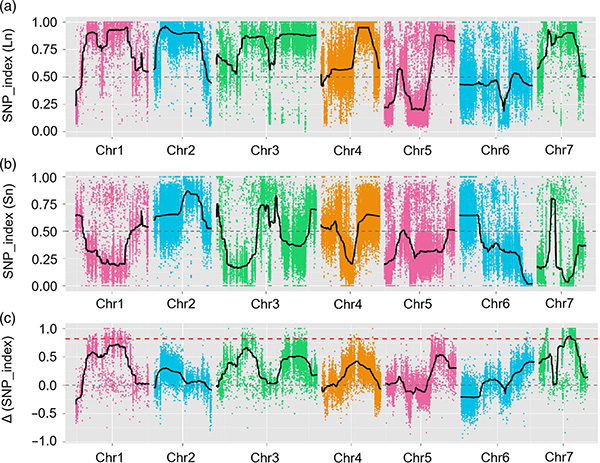 Mapping QTL na análise BSA para identificar a rexión candidata asociada á lonxitude do pescozo do pepino | 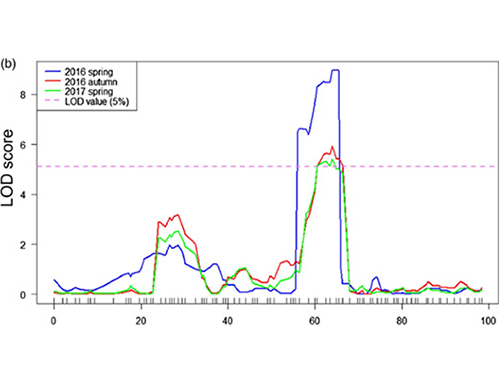 Perfís LOD de qtl de pescozo de pepino identificado en Chr07 |
Xu, X., et al. "O trazo cuantitativo de gran efecto FNL7.1 codifica unha abundante proteína de embrioxénese asociada á lonxitude do pescozo do pepino." Plant Biotechnology Journal 18.7 (2020).



