

Transcriptome spatial BMKMANU S1000

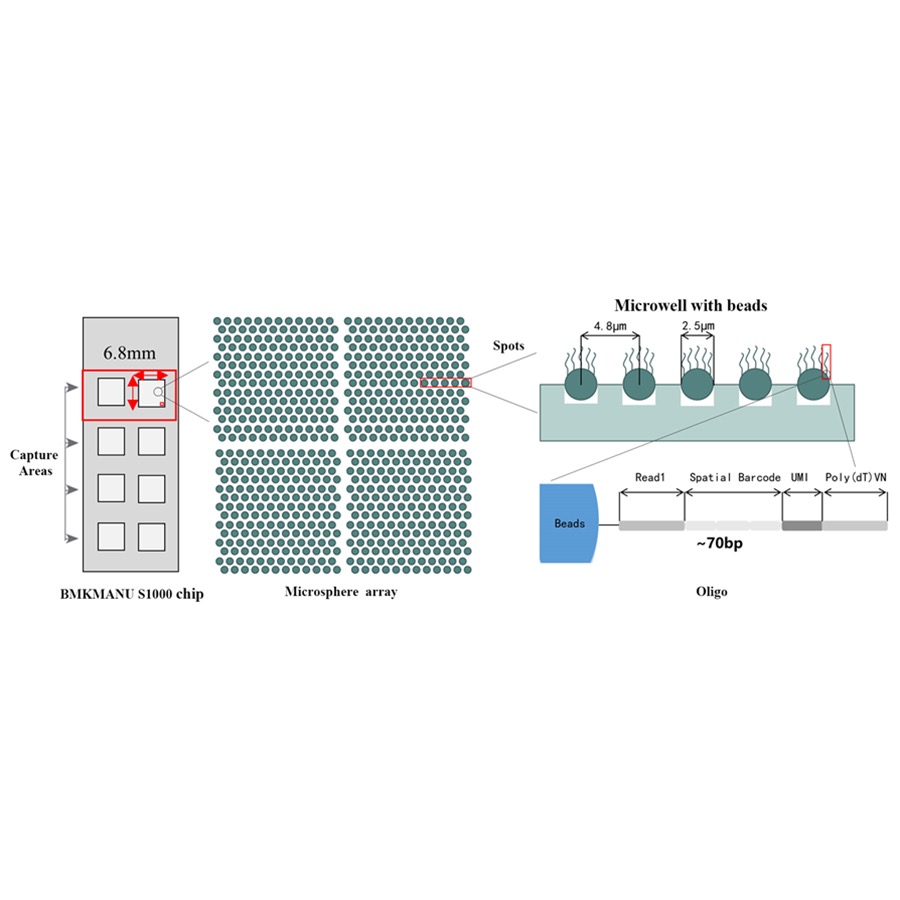
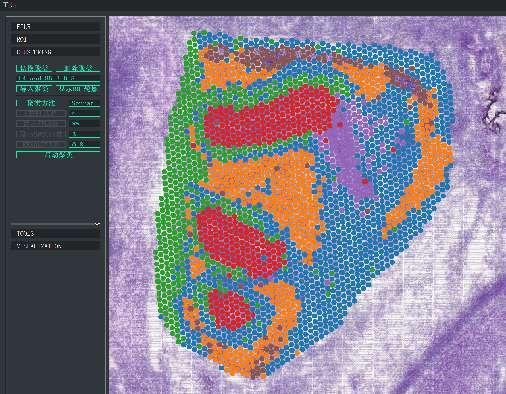
Schéma technique du transcriptome spatial BMKMANU S1000

Caractéristiques
● Résolution : 5 µM
● Diamètre du point : 2,5 µM
● Nombre de spots : environ 2 millions
● 3 formats de zone de capture possibles : 6,8 mm * 6,8 mm, 11 mm * 11 mm ou 15 mm * 20 mm
● Chaque bille à code-barres est chargée d'amorces composées de 4 sections :
queue poly (dT) pour l'amorçage de l'ARNm et la synthèse de l'ADNc
Identifiant moléculaire unique (UMI) pour corriger le biais d'amplification
Code-barres spatial
Séquence de liaison de l'amorce de séquençage à lecture partielle 1
● Coloration H&E et fluorescente des coupes
● Possibilité d'utilisertechnologie de segmentation cellulaire: intégration de la coloration H&E, de la coloration fluorescente et du séquençage de l'ARN pour déterminer les limites de chaque cellule et attribuer correctement l'expression des gènes à chaque cellule.
Avantages du BMKMANU S1000
●Résolution sous-cellulaire: Chaque zone de capture contenait plus de 2 millions de spots spatiaux codés avec un diamètre de 2,5 µm et un espacement de 5 µm entre les centres des spots, permettant une analyse spatiale du transcriptome avec une résolution subcellulaire (5 µm).

●Analyse de résolution multi-niveaux :Analyse flexible à plusieurs niveaux allant de 100 μm à 5 μm pour résoudre diverses caractéristiques des tissus avec une résolution optimale.

●Possibilité d'utiliser la technologie de segmentation cellulaire « Trois en une » :Combinant la coloration par fluorescence, la coloration H&E et le séquençage de l'ARN sur une seule lame, notre algorithme d'analyse « trois en un » permet l'identification des limites cellulaires pour une transcriptomique cellulaire ultérieure.

●Compatible avec plusieurs plates-formes de séquençage: Séquençage NGS et lecture longue disponible.
●Conception flexible de 1 à 8 zones de capture actives: La taille de la zone de capture est flexible, pouvant utiliser 3 formats (6,8 mm * 6,8 mm., 11 mm * 11 mm et 15 mm * 20 mm)
●Service à guichet unique: Il intègre toutes les étapes basées sur l'expérience et les compétences, y compris la cryosection, la coloration, l'optimisation des tissus, le codage à barres spatial, la préparation de bibliothèques, le séquençage et la bioinformatique.
●Bioinformatique complète et visualisation conviviale des résultats :Le package comprend 29 analyses et plus de 100 chiffres de haute qualité, combinés à l’utilisation d’un logiciel développé en interne pour visualiser et personnaliser la division cellulaire et le regroupement ponctuel.
●Analyse et visualisation de données personnalisées: disponible pour différentes demandes de recherche
●Une équipe technique hautement qualifiée: avec une expérience dans plus de 250 types de tissus et plus de 100 espèces, dont l'homme, la souris, les mammifères, les poissons et les plantes.
●Mises à jour en temps réel sur l'ensemble du projet: avec un contrôle total des progrès expérimentaux.
●Analyse conjointe facultative avec séquençage d’ARNm unicellulaire
Spécifications des services
|
Échantillon Exigences
| Bibliothèque |
Stratégie de séquençage
| Données recommandées | Contrôle de qualité |
| Échantillons cryo intégrés à l'OCT, 3 blocs par échantillon | Bibliothèque d'ADNc S1000 | Illumina PE150 (autres plateformes disponibles) | 100 000 lectures PE pour 100 µM ( 60-150 Go ) | RIN>7 |
Pour plus de détails sur les conseils de préparation des échantillons et le flux de travail du service, n'hésitez pas à parler à unExpert BMKGENE
Flux de travail des services
Au cours de la phase de préparation des échantillons, un premier essai d’extraction d’ARN en vrac est effectué pour garantir l’obtention d’un ARN de haute qualité. Au cours de l’étape d’optimisation des tissus, les coupes sont colorées et visualisées et les conditions de perméabilisation pour la libération de l’ARNm des tissus sont optimisées. Le protocole optimisé est ensuite appliqué lors de la construction de la bibliothèque, suivi du séquençage et de l'analyse des données.
Le flux de travail complet du service implique des mises à jour en temps réel et des confirmations des clients pour maintenir une boucle de rétroaction réactive, garantissant ainsi une exécution fluide du projet.


Les données générées par BMKMANU S1000 sont analysées à l'aide du logiciel « BSTMatrix », conçu indépendamment par BMKGENE, générant une matrice d'expression génique. À partir de là, un rapport standard est généré qui comprend le contrôle de la qualité des données, l’analyse de l’échantillon interne et l’analyse inter-groupes.
● Contrôle de la qualité des données :
Sortie des données et distribution du score de qualité
Détection de gènes par spot
Couverture des tissus
● Analyse d'échantillon interne :
Richesse génétique
Regroupement ponctuel, y compris l'analyse de dimensions réduites
Analyse d'expression différentielle entre clusters : identification de gènes marqueurs
Annotation fonctionnelle et enrichissement de gènes marqueurs
● Analyse inter-groupes :
Recombinaison des spots des deux échantillons (par exemple malades et contrôle) et re-cluster
Identification des gènes marqueurs pour chaque cluster
Annotation fonctionnelle et enrichissement de gènes marqueurs
Expression différentielle du même cluster entre les groupes
De plus, BMKGENE a développé « BSTViewer » qui est un outil convivial qui permet à l'utilisateur de visualiser l'expression des gènes et de repérer le regroupement à différentes résolutions.
Logiciel développé par BMKGene pour une visualisation conviviale
Clustering ponctuel BSTViewer à résolution multi-niveaux

BSTCellViewer : fractionnement de cellules automatique et manuel

Analyse d'échantillon interne
Regroupement ponctuel :

Identification des gènes marqueurs et répartition spatiale :

Analyse inter-groupes
Combinaison de données des deux groupes et du reclustage :

Gènes marqueurs de nouveaux clusters :

Découvrez les progrès facilités par les services de transcriptomique spatiale de BMKGene avec la technologie BMKManu S1000 dans cette publication vedette :
Chanson, X. et al. (2023) « La transcriptomique spatiale révèle des cellules de chlorenchyme induites par la lumière impliquées dans la promotion de la régénération des pousses dans les cals de la tomate »,Actes de l'Académie nationale des sciences des États-Unis d'Amérique, 120(38), p. e2310163120. est ce que je: 10.1073/pnas.2310163120
Vous, Y. et al. (2023) 'Comparaison systématique des méthodes de transcriptomique spatiale basées sur le séquençage',bioRxiv, p. 2023.12.03.569744. est ce que je: 10.1101/2023.12.03.569744.






{kind=link}