

BMKMANU S1000 Spatial Transcriptome

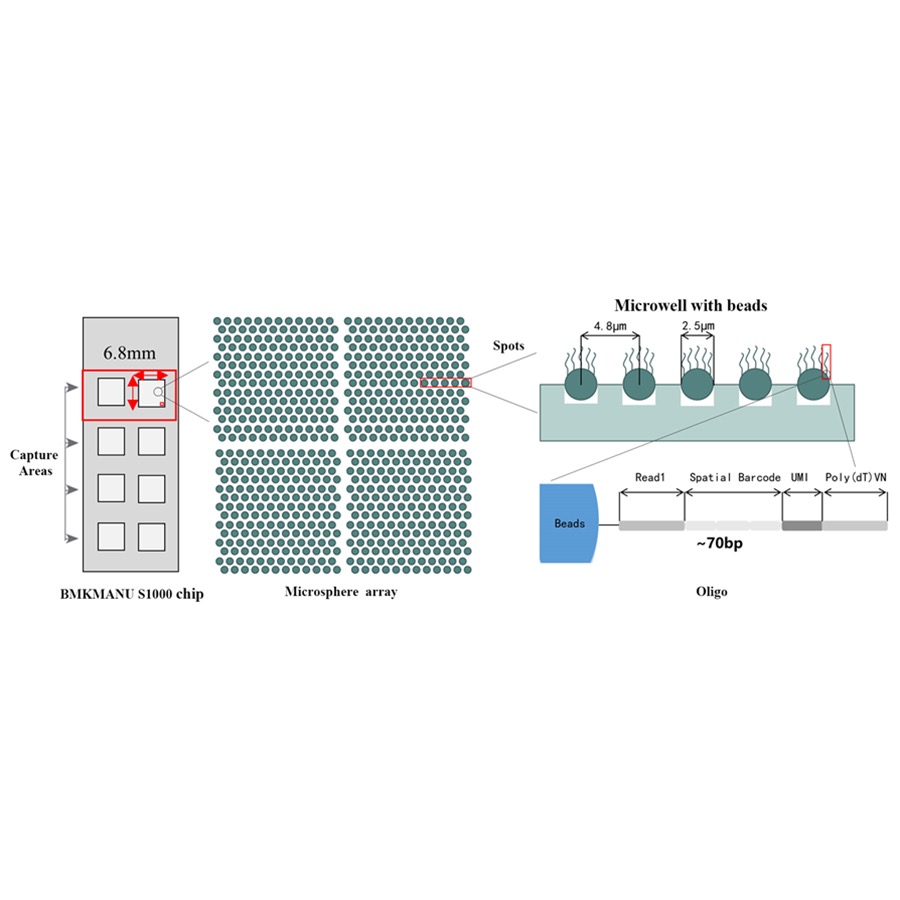
BMKMANU S1000 Spatial Transcriptome Technical Scheme

Ominaisuudet
● Resoluutio: 5 µM
● Pistehalkaisija: 2,5 µM
● Kohteiden määrä: noin 2 miljoonaa
● 3 mahdollista kuvausalueen muotoa: 6,8 mm * 6,8 mm, 11 mm * 11 mm tai 15 mm * 20 mm
● Jokainen viivakoodihelmi on ladattu pohjamaaleilla, jotka koostuvat 4 osasta:
poly(dT)-häntä mRNA-alukkeelle ja cDNA-synteesiin
Unique Molecular Identifier (UMI) vahvistaa amplifikaatiota
Spatiaalinen viivakoodi
Osittain luetun 1 sekvensointialukkeen sitoutumissekvenssi
● Leikkeiden H&E ja fluoresoiva värjäys
● Mahdollisuus käyttääsolusegmentointitekniikka: H&E-värjäyksen, fluoresoivan värjäyksen ja RNA-sekvensoinnin integrointi kunkin solun rajojen määrittämiseksi ja geenin ilmentymisen osoittamiseksi oikein kullekin solulle.
BMKMANU S1000:n edut
●Subcellular resoluutio: Kukin sieppausalue sisälsi > 2 miljoonaa spatiaalista viivakoodattua täplää, joiden halkaisija oli 2,5 µm ja 5 µm:n etäisyys täpläkeskuksien välillä, mikä mahdollisti spatiaalisen transkriptomianalyysin solun alaresoluutiolla (5 µm).

●Monitasoinen resoluutioanalyysi:Joustava monitasoinen analyysi 100 μm:stä 5 μm:iin erilaisten kudosten ominaisuuksien ratkaisemiseksi optimaalisella resoluutiolla.

●Mahdollisuus käyttää "Kolme yhdessä diassa" -solusegmentointitekniikkaa:Yhdistämällä fluoresenssivärjäyksen, H&E-värjäyksen ja RNA-sekvensoinnin yhdelle dialle, "kolme yhdessä" -analyysialgoritmimme mahdollistaa solurajojen tunnistamisen myöhempää solupohjaista transkriptomiikkaa varten.

●Yhteensopiva useiden sekvensointialustojen kanssa: Saatavilla on sekä NGS- että pitkälukuinen sekvensointi.
●Joustava 1-8 aktiivisen kuvausalueen muotoilu: Kuvausalueen koko on joustava, ja siinä on mahdollista käyttää 3 muotoa (6,8 mm * 6,8 mm., 11 mm * 11 mm ja 15 mm * 20 mm)
●Yhden luukun palvelu: Se yhdistää kaikki kokemukseen ja taitoihin perustuvat vaiheet, mukaan lukien kryoleikkaus, värjäys, kudosten optimointi, spatiaalinen viivakoodaus, kirjaston valmistelu, sekvensointi ja bioinformatiikka.
●Kattava bioinformatiikka ja käyttäjäystävällinen tulosten visualisointi:Paketti sisältää 29 analyysiä ja yli 100 korkealaatuista lukua yhdistettynä sisäiseen ohjelmistoon solujen jakamisen ja spot-klusteroinnin visualisointiin ja mukauttamiseen.
●Räätälöity tietojen analysointi ja visualisointi: saatavilla erilaisiin tutkimuspyyntöihin
●Korkeasti koulutettu tekninen tiimi: kokemusta yli 250 kudostyypistä ja yli 100 lajista, mukaan lukien ihminen, hiiri, nisäkäs, kala ja kasvi.
●Reaaliaikaiset päivitykset koko projektista: kokeen edistymisen täysi hallinta.
●Valinnainen yhteisanalyysi yksisoluisen mRNA-sekvensoinnin avulla
Palvelun tiedot
|
Näyte Vaatimukset
| Kirjasto |
Sekvensointistrategia
| Tietoja suositellaan | Laadunvalvonta |
| OCT-upotetut kryonäytteet, 3 lohkoa näytettä kohti | S1000 cDNA-kirjasto | Illumina PE150 (muita alustoja saatavilla) | 100K PE lukemaa 100 uM:aa kohti (60-150 Gt) | RIN>7 |
Saat lisätietoja näytteiden valmistelun ohjeista ja palvelun työnkulusta ottamalla yhteyttä aBMKGENE asiantuntija
Palvelun työnkulku
Näytteen valmistusvaiheessa suoritetaan ensimmäinen bulkki-RNA:n uuttokoe, jotta voidaan varmistaa korkealaatuisen RNA:n saaminen. Kudosoptimointivaiheessa leikkeet värjätään ja visualisoidaan ja permeabilisaatioolosuhteet mRNA:n vapautumiselle kudoksesta optimoidaan. Optimoitua protokollaa sovelletaan sitten kirjaston rakentamisen aikana, minkä jälkeen suoritetaan sekvensointi ja data-analyysi.
Täydellinen palvelun työnkulku sisältää reaaliaikaiset päivitykset ja asiakasvahvistukset responsiivisen palautesilmukan ylläpitämiseksi, mikä varmistaa projektin sujuvan toteutuksen.


BMKMANU S1000:n tuottamat tiedot analysoidaan BMKGENE:n itsenäisesti suunnittelemalla "BSTMatrix" -ohjelmistolla, joka tuottaa Gene Expression Matrixin. Sieltä luodaan vakioraportti, joka sisältää tietojen laadunvalvonnan, sisäisen otosanalyysin ja ryhmien välisen analyysin.
● Tietojen laadunvalvonta:
Tiedon tuotto ja laatupisteiden jakautuminen
Geenitunnistus per kohta
Kudosten peitto
● Sisäinen otosanalyysi:
Geenien rikkaus
Pisteklusterointi, mukaan lukien pienennetyn ulottuvuuden analyysi
Differentiaalinen ilmentymisanalyysi klustereiden välillä: markkerigeenien tunnistaminen
Markkerigeenien toiminnallinen annotaatio ja rikastaminen
● Ryhmien välinen analyysi:
Molempien näytteiden (esim. sairaiden ja kontrollien) täplien yhdistäminen ja ryhmittyminen uudelleen
Merkkigeenien tunnistaminen jokaiselle klusterille
Markkerigeenien toiminnallinen annotaatio ja rikastaminen
Saman klusterin differentiaalinen ilmaisu ryhmien välillä
Lisäksi BMKGENE:n kehittämä ”BSTViewer” on käyttäjäystävällinen työkalu, jonka avulla käyttäjä voi visualisoida geenien ilmentymisen ja spot-klusteroinnin eri resoluutioilla.
BMKGene kehitti ohjelmiston käyttäjäystävälliseen visualisointiin
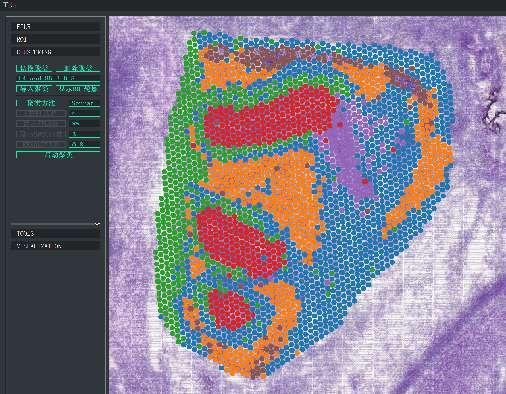
BSTViewer-pisteklusterointi monitasoisella resoluutiolla

BSTCellViewer: automaattinen ja manuaalinen solujen jakaminen

Sisäisen näytteen analyysi
Pisteklusterointi:

Markkerigeenien tunnistus ja tilajakauma:

Ryhmien välinen analyysi
Tietojen yhdistelmä molemmista ryhmistä ja uudelleenklusterista:

Uusien klustereiden markkerigeenit:

Tutustu BMKGenen tilatranskriptomiikan ja BMKManu S1000 -teknologian edistämiin edistysaskeliin tässä esitellyssä julkaisussa:
Song, X. et ai. (2023) "Spatiaalinen transkriptomiikka paljastaa valon aiheuttamia klorenkyymasoluja, jotka edistävät versojen regeneraatiota tomaattikalluksessa",Proceedings of the National Academy of Sciences of the United of America, 120(38), s. e2310163120. doi: 10.1073/pnas.2310163120
Sinä, Y. et ai. (2023) "Sekvensointiin perustuvien spatiaalisten transkriptomisten menetelmien järjestelmällinen vertailu",bioRxiv, s. 2023.12.03.569744. doi: 10.1101/2023.12.03.569744.






{kind=link}