

Secuenciación de fragmentos amplificados de locos específico (SLAF-seq)

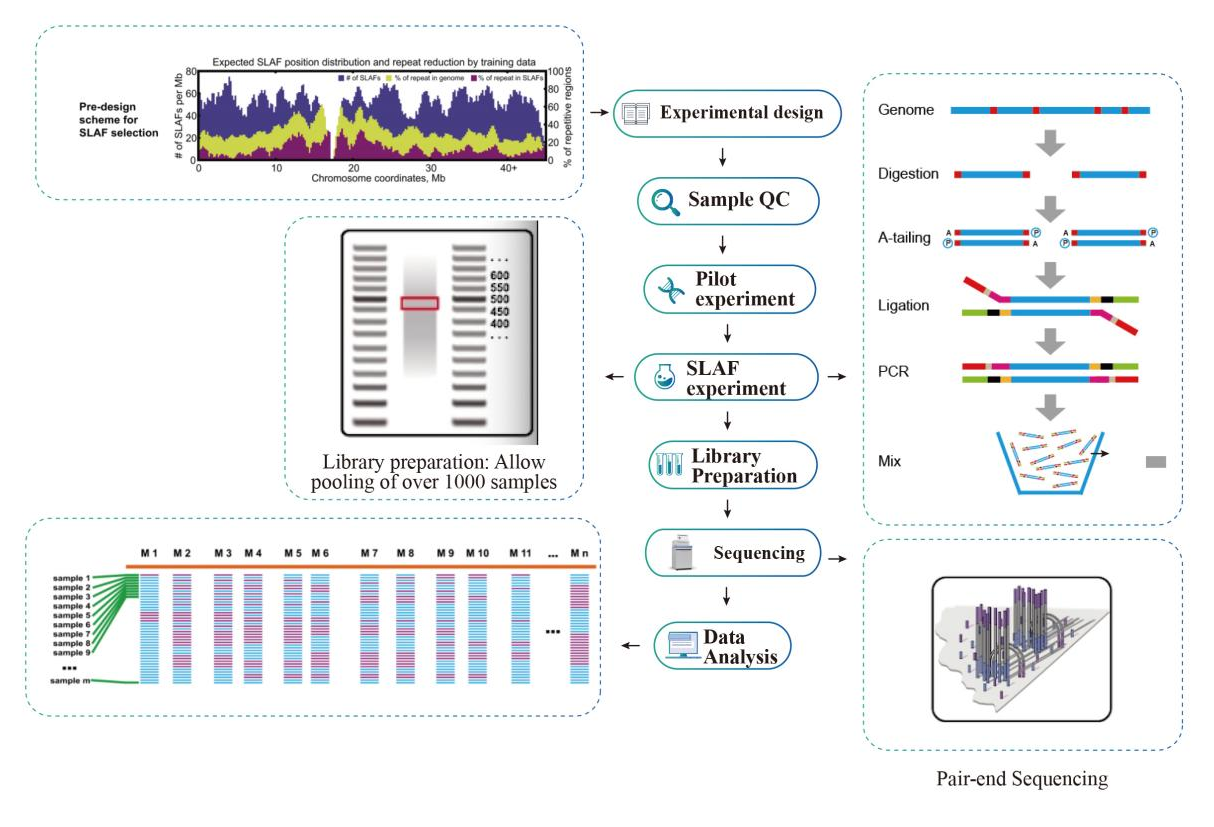
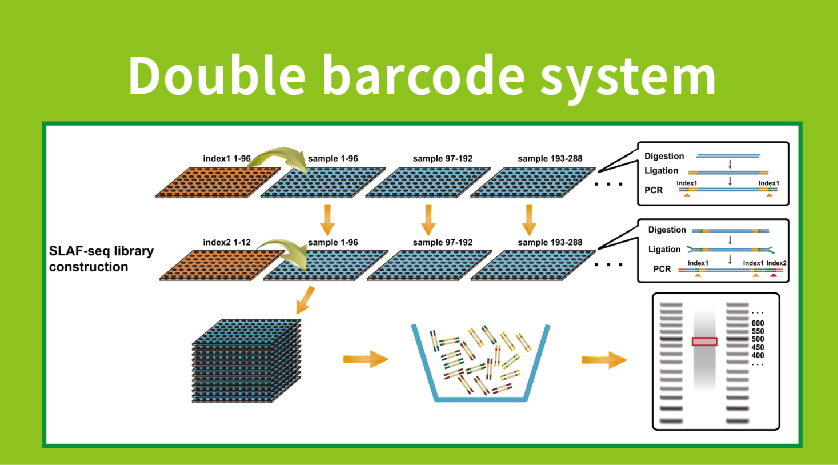
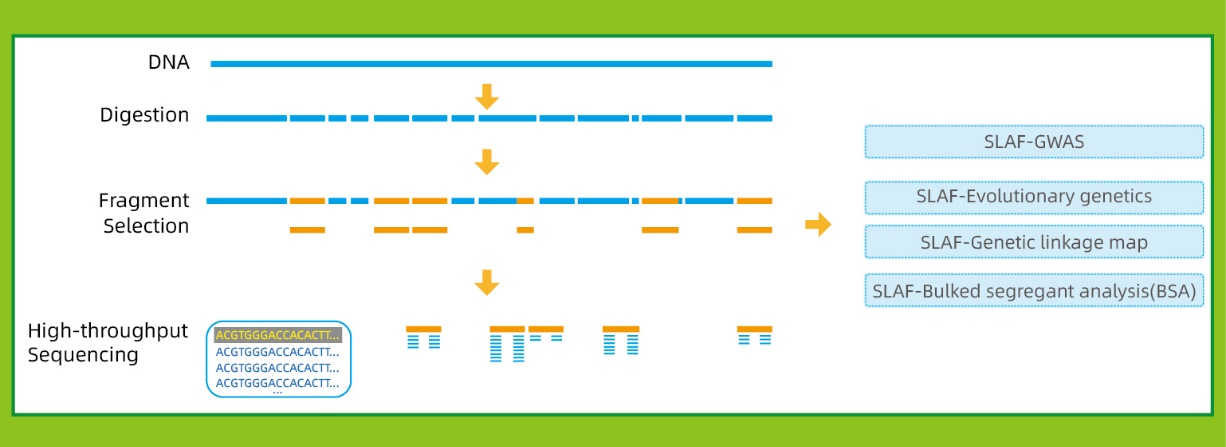
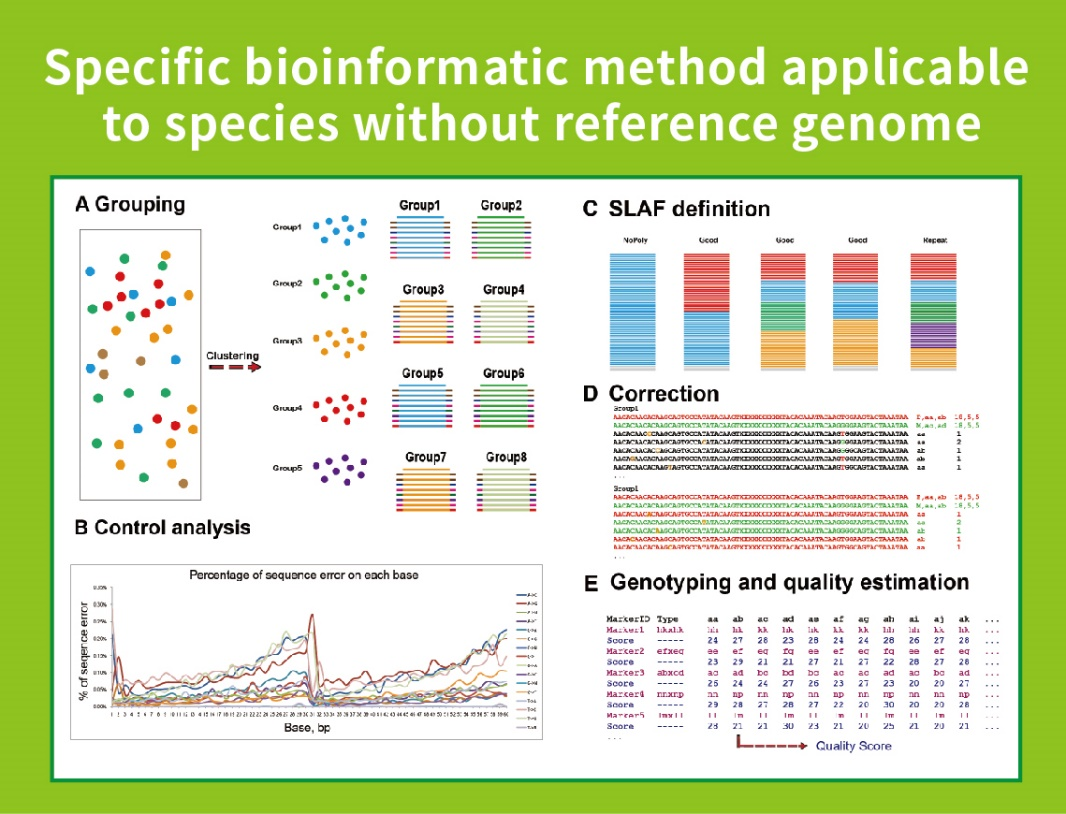
Flujo de trabajo

Esquema técnico

Características de servicio
● Secuenciación en NovaseQ con PE150.
● Preparación de la biblioteca con código de barras doble, lo que permite la agrupación de más de 1000 muestras.
● Esta técnica se puede usar con o sin un genoma de referencia, con diferentes tuberías bioinformáticas para cada caso:
Con genoma de referencia: SNP e Indel Discovery
Sin genoma de referencia: agrupación de muestras y descubrimiento de SNP
● En elen silicoLas combinaciones de enzimas de restricción múltiple de la etapa previa al diseño se detectan para encontrar las que generan una distribución uniforme de etiquetas SLAF a lo largo del genoma.
● Durante el preexperimento, se prueban tres combinaciones de enzimas en 3 muestras para generar 9 bibliotecas SLAF, y esta información se usa para elegir la combinación de enzimas de restricción óptima para el proyecto.
Ventajas de servicio
●Descubrimiento de marcadores genéticos altos: La integración de un sistema de código de barras doble de alto rendimiento permite la secuenciación simultánea de grandes poblaciones, y la amplificación específica del locus mejora la eficiencia, asegurando que los números de etiquetas cumplan con los diversos requisitos de varias preguntas de investigación.
● Baja dependencia del genoma: Se puede aplicar a especies con o sin un genoma de referencia.
●Diseño de esquema flexible: Enzima única, enzima dual, digestión de enzima múltiple y varios tipos de enzimas pueden seleccionarse para atender diferentes objetivos o especies de investigación. Elen silicoEl diseño previo se lleva a cabo para garantizar un diseño de enzimas óptimo.
● Alta eficiencia en la digestión enzimática: La conducción de unen silicoEl diseño previo y un diseño óptimo previo a la experiencia aseguraron una distribución uniforme de etiquetas SLAF en el cromosoma (1 etiqueta SLAF/4KB) y una secuencia repetitiva reducida (<5%).
●Experiencia extensa: Nuestro equipo aporta una gran experiencia a cada proyecto, con un historial de cierre de más de 5000 proyectos SLAF-seq en cientos de especies, incluidas plantas, mamíferos, aves, insectos y organismos acuáticos.
● Flujo de trabajo bioinformático autodesarrollado: BMKGene desarrolló un flujo de trabajo bioinformático integrado para SLAF-seq para garantizar la confiabilidad y precisión de la salida final.
Especificaciones de servicio
| Tipo de análisis | Escala de población recomendada | Estrategia de secuenciación | |
| Profundidad de secuenciación de etiquetas | Número de etiqueta | ||
| Mapas genéticos | 2 padres y> 150 descendientes | Padres: 20x WGS Offsping: 10x | Tamaño del genoma: <400 MB: se recomienda WGS <1GB: 100k etiquetas 1-2GB :: 200k Etiquetas > 2GB: 300k etiquetas Max 500k Etiquetas |
| Estudios de asociación de todo el genoma (GWAS) | ≥200 muestras | 10x | |
| Evolución genética | ≥30 muestras, con> 10 muestras de cada subgrupo | 10x | |
Requisitos de servicio
Concentración ≥ 5 ng/µl
Cantidad total ≥ 80 ng
Nanodrop OD260/280 = 1.6-2.5
Gel de agarosa: degradación o contaminación no o limitada
Entrega de muestra recomendada
Contenedor: Tubo de centrífuga de 2 ml
(Para la mayoría de las muestras, recomendamos no preservar en etanol)
Etiquetado de muestra: las muestras deben etiquetarse claramente e idénticas al formulario de información de muestra enviado.
Envío: hielo seco: las muestras deben embalarse primero en bolsas y enterradas en hielo seco.
Flujo de trabajo de servicio






Muestra de control de calidad
Experimento piloto
Experimento
Preparación de la biblioteca
Secuenciación
Análisis de datos
Servicios posteriores
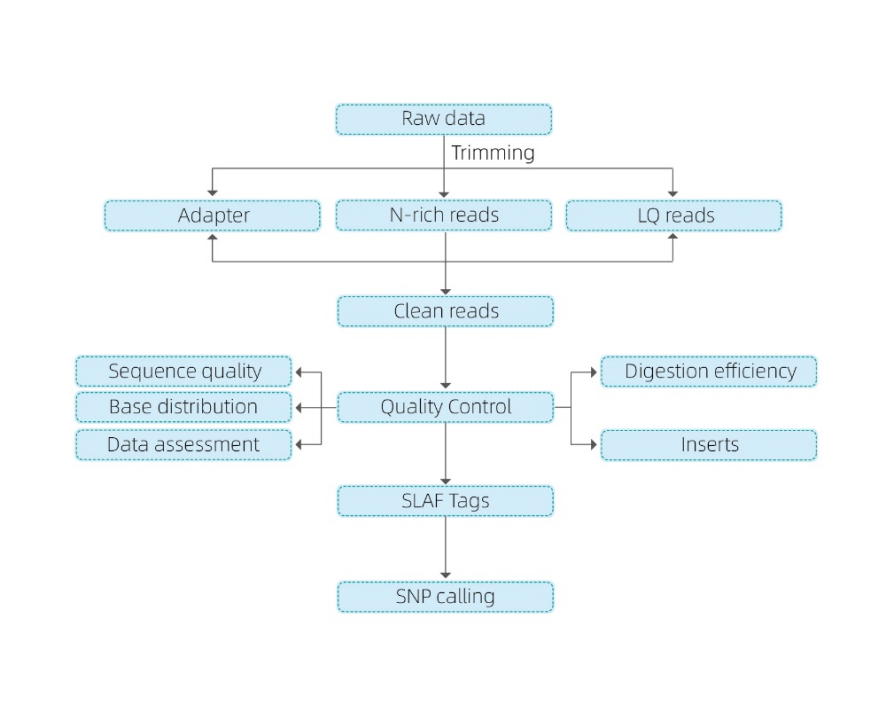 Incluye el siguiente análisis:
Incluye el siguiente análisis:

- Secuenciación de datos QC
- Desarrollo de etiquetas SLAF
Mapeo para hacer referencia al genoma
Sin un genoma de referencia: agrupación
- Análisis de etiquetas SLAF.: Estadísticas, distribución de todo el genoma
- Descubrimiento de marcadores: SNP, Indel, SNV, CV llamadas y anotación
Distribución de etiquetas SLAF en cromosomas:

Distribución de SNP en los cromosomas:
 Anotación SNP
Anotación SNP

| Año | Diario | IF | Título | Aplicaciones |
| 2022 | Comunicaciones de la naturaleza | 17.694 | Base genómica de los cromosomas Giga y Giga de la peonía de los árboles Paeonia Ostii | Slaf-gwas |
| 2015 | Nuevo fitólogo | 7.433 | Las huellas de domesticación anclan regiones genómicas de importancia agronómica en soja | Slaf-gwas |
| 2022 | Revista de investigación avanzada | 12.822 | Introgresiones artificiales de todo el genoma de Gossypium Barbadense en G. hirsutum Revelar loci superiores para la mejora simultánea de la calidad y rendimiento de la fibra de algodón rasgos | Genética evolutiva de slaf |
| 2019 | Planta molecular | 10.81 | El análisis genómico de la población y el ensamblaje de novo revelan el origen de la maleza Arroz como juego evolutivo | Genética evolutiva de slaf |
| 2019 | Genética de la naturaleza | 31.616 | Secuencia del genoma y diversidad genética de la carpa común, Cyprinus carpio | Mapa de enlace slaf |
| 2014 | Genética de la naturaleza | 25.455 | El genoma del maní cultivado proporciona información sobre los cariotipos de legumbres, poliploides Evolución y domesticación de cultivos. | Mapa de enlace slaf |
| 2022 | Revista de biotecnología vegetal | 9.803 | La identificación de ST1 revela una selección que involucra autostopistas de la morfología de las semillas y contenido de aceite durante la domesticación de la soja | Desarrollo de marcadores SLAF |
| 2022 | Revista Internacional de Ciencias Moleculares | 6.208 | Identificación y desarrollo de marcadores de ADN para un trigo-leymus mollis 2ns (2d) Sustitución del cromosoma disómico | Desarrollo de marcadores SLAF |
| Año | Diario | IF | Título | Aplicaciones |
| 2023 | Fronteras en ciencias de las plantas | 6.735 | Mapeo QTL y análisis del transcriptoma del contenido de azúcar durante la maduración de la fruta de Pyrus Pyrifolia | Mapa genético |
| 2022 | Revista de biotecnología vegetal | 8.154 | La identificación de ST1 revela una selección que implica autostopear la morfología de las semillas y el contenido de aceite durante la domesticación de la soja
| Llamadas SNP |
| 2022 | Fronteras en ciencias de las plantas | 6.623 | Mapeo de la asociación de todo el genoma de los fenotipos de cavos apenas en el entorno de sequía.
| Gwas |





