

Genómica comparada

Ventajas del servicio
●Amplia experiencia y registros de publicaciones.: con lo acumulado, BMKGene ha completado más de 90 proyectos de genómica comparativa, con un factor de impacto acumulado de 900.
●Análisis bioinformático integral.: el paquete de análisis contiene los ocho análisis más comúnmente requeridos, proporciona cifras bien diseñadas listas para publicar y permite una fácil interpretación de los resultados.
●Equipo bioinformático altamente cualificado y ciclo de análisis corto.: con gran experiencia en análisis genómico comparativo, el equipo de BMKGene satisface diversas demandas de análisis personalizados en un corto tiempo de respuesta.
●Soporte Postventa:Nuestro compromiso se extiende más allá de la finalización del proyecto con un período de servicio postventa de 3 meses. Durante este tiempo, ofrecemos seguimiento del proyecto, asistencia para la resolución de problemas y sesiones de preguntas y respuestas para abordar cualquier consulta relacionada con los resultados.
Especificaciones de servicio
| Tiempo estimado de respuesta | Número de especies | Análisis |
| 30 días laborables | 6 - 12 | Agrupación de familias de genes Expansión y contracción de la familia genética. Construcción de árboles filogenéticos. Estimación del tiempo de divergencia (se requiere calibración fósil) Tiempo de inserción LTR (Para plantas) Duplicación del genoma completo (para plantas) Presión selectiva Análisis de sintenia |
Análisis bioinformáticos.
● Familia de genes
● Filogenética
● Tiempo de divergencia
● Presión selectiva
● Análisis de sintenia
Requisitos de muestra y entrega
Requisitos de muestra:
Tejido o ADN para secuenciación y ensamblaje del genoma.
Para tejido
| Especies | Tejido | Encuesta | PacBio CCS |
| Animal | tejido visceral | 0,5 ~ 1 gramos | ≥ 3,5 gramos |
| Tejido muscular | |||
| ≥ 5,0 gramos | |||
| ≥ 5,0 ml | |||
| sangre de mamífero | |||
| ≥ 0,5 ml | |||
| Sangre de aves/pescado | |||
| Planta | hoja fresca | 1 ~ 2 gramos | ≥ 5,0 gramos |
| Pétalo/tallo | 1 ~ 2 gramos | ≥ 10,0 gramos | |
| Raíz/Semilla | 1 ~ 2 gramos | ≥ 20,0 gramos | |
| Células | Célula cultivada | - | ≥ 1 x 108 |
Datos
Archivos de secuencia del genoma (.fasta) y archivos de anotaciones (.gff3) de especies estrechamente relacionadas
Flujo de trabajo del servicio

Diseño de experimentos

Entrega de muestra

construcción de biblioteca

Secuenciación

Análisis de datos

Servicios postventa
*Los resultados de demostración que se muestran aquí provienen todos de genomas publicados con Biomarker Technologies.
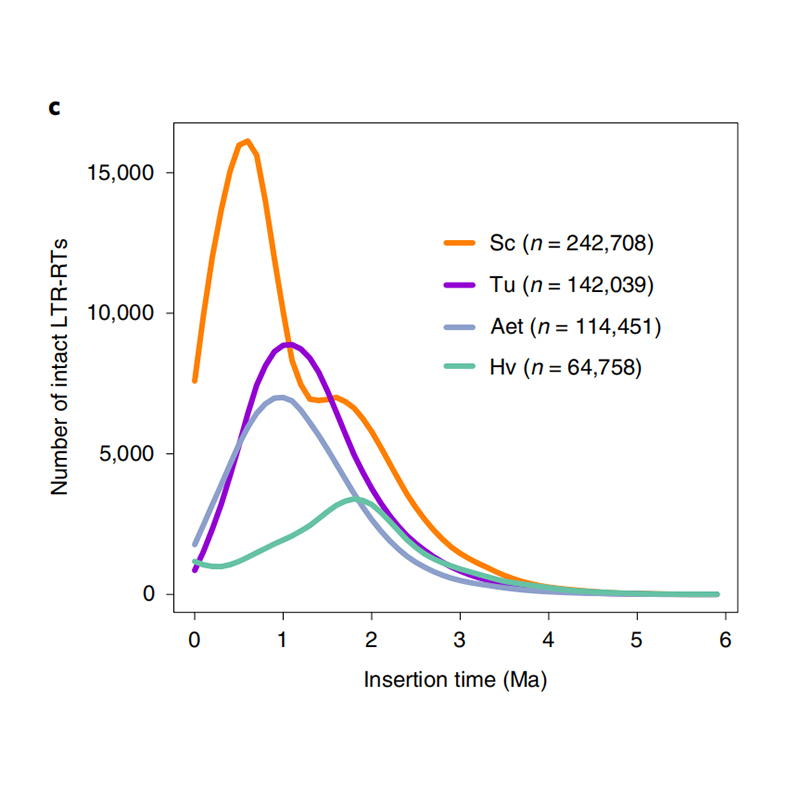
1.Estimación del tiempo de inserción de LTR: la figura muestra una distribución bimodal única en los tiempos de inserción de LTR-RT en el genoma del centeno Weining, en comparación con otras especies. El pico más reciente apareció hace alrededor de 0,5 millones de años.
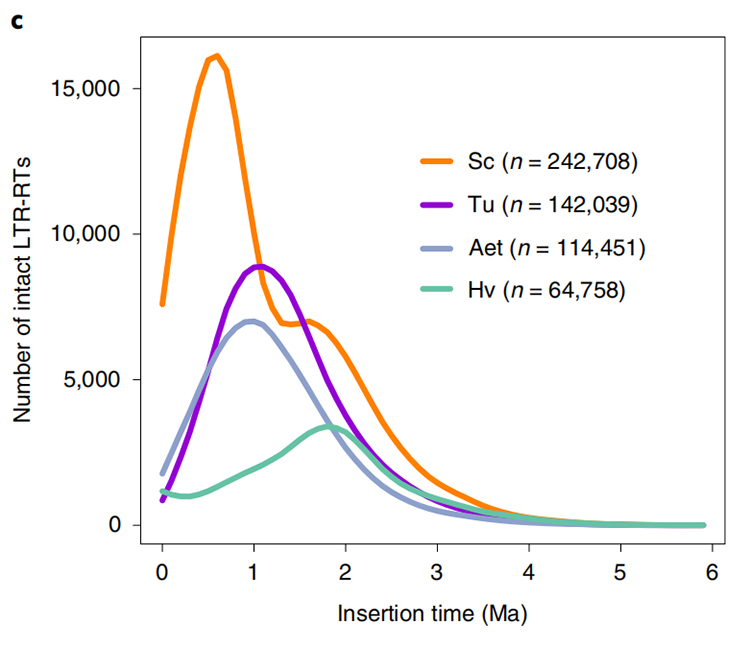
Li Guang et al.,Genética de la naturaleza, 2021
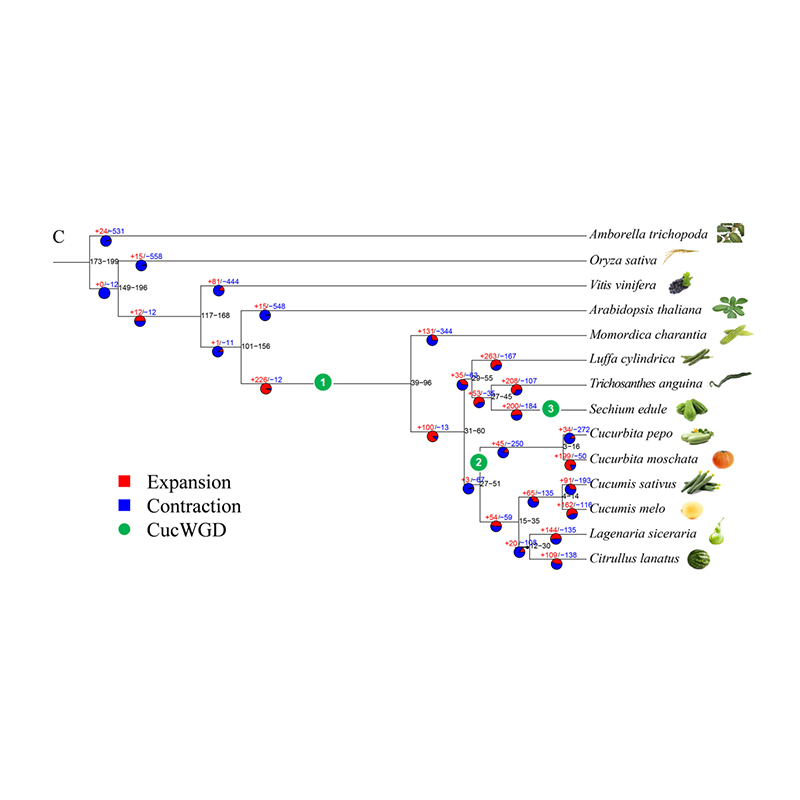
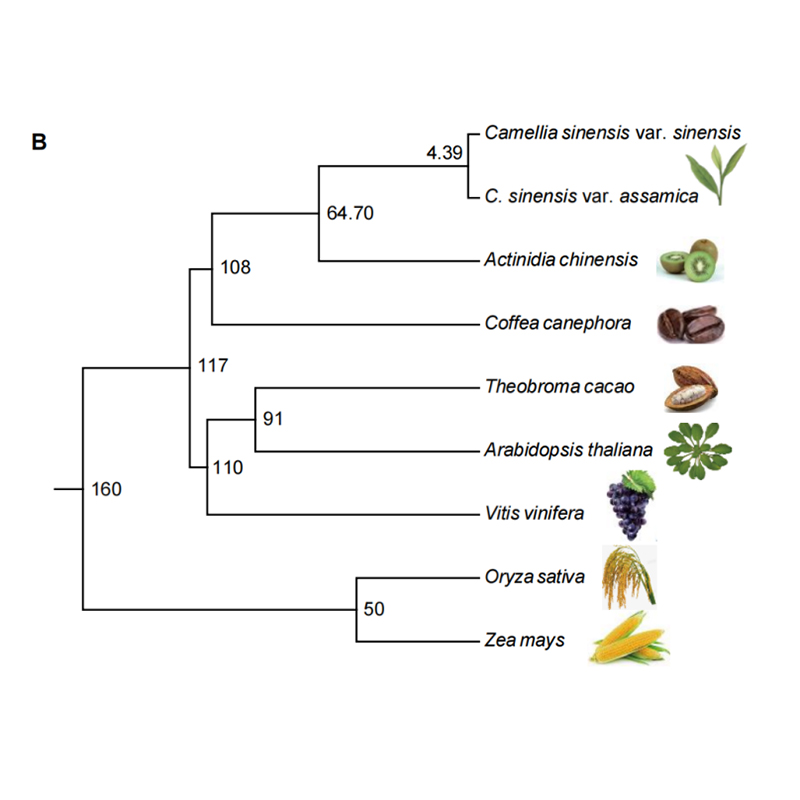
2. Análisis de filogenia y familia de genes del chayote (Sechium edule): Al analizar el chayote y las otras 13 especies relacionadas en la familia de genes, se descubrió que el chayote está más estrechamente relacionado con la calabaza serpiente (Trichosanthes anguina). Chayote derivado de calabaza serpiente en alrededor de 27-45 millones de años y se observó duplicación del genoma completo (WGD) en chayote en 25 ± 4 millones de años, que es el tercer evento de WGD en cucuibitaceae.
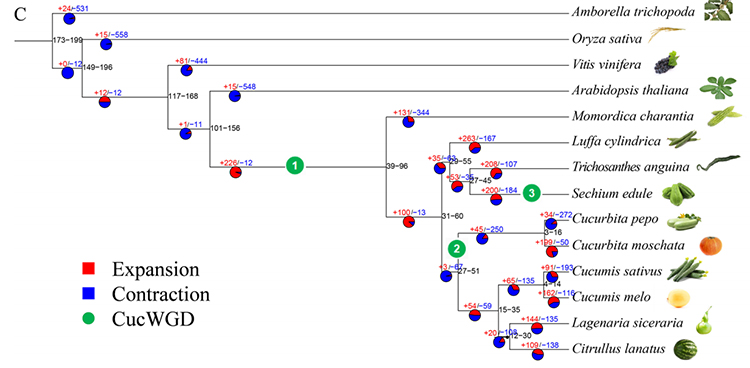
Fu A y otros,Investigación en horticultura, 2021
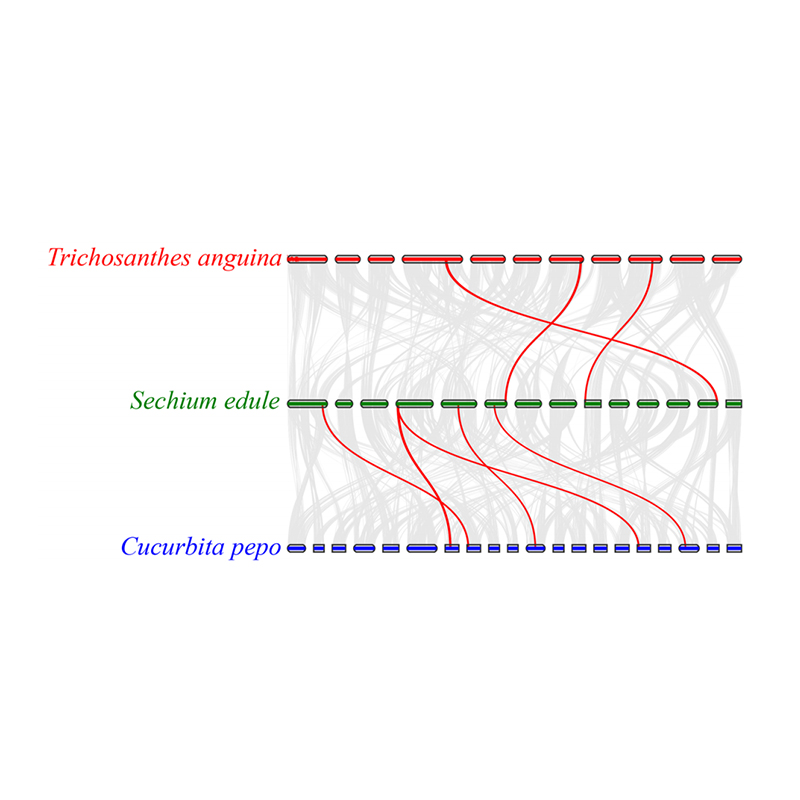
3.Análisis de síntesis: Se encontraron algunos genes relacionados con fitohormonas en el desarrollo del fruto en chayote, calabaza serpiente y calabaza. La correlación entre chayote y calabaza es ligeramente mayor que entre chayote y calabaza serpiente.
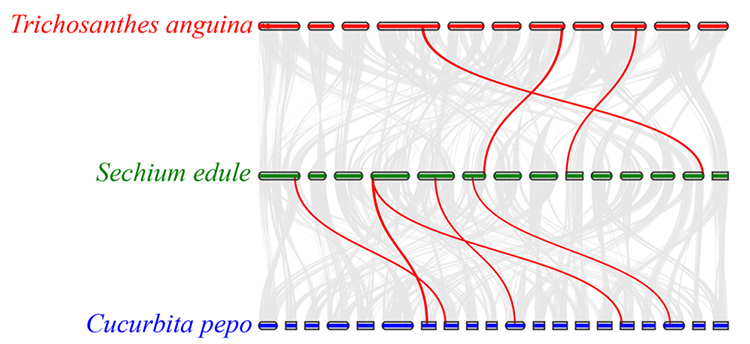
Fu A y otros,Investigación en horticultura, 2021
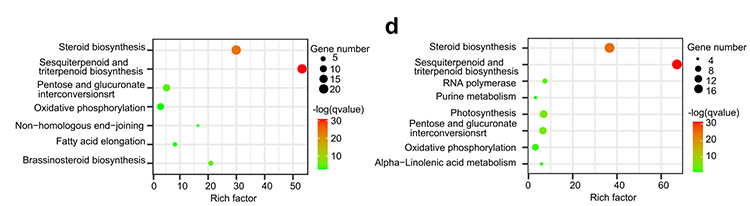
4.Análisis de la familia de genes: el enriquecimiento con KEGG sobre la expansión y contracción de la familia de genes en los genomas de G.thurberi y G.davidsonii mostró que se expandieron los genes relacionados con la biosíntesis de esteroides y la biosíntesis de brasinoesteroides.
Yang Z y otros,Biología BMC, 2021
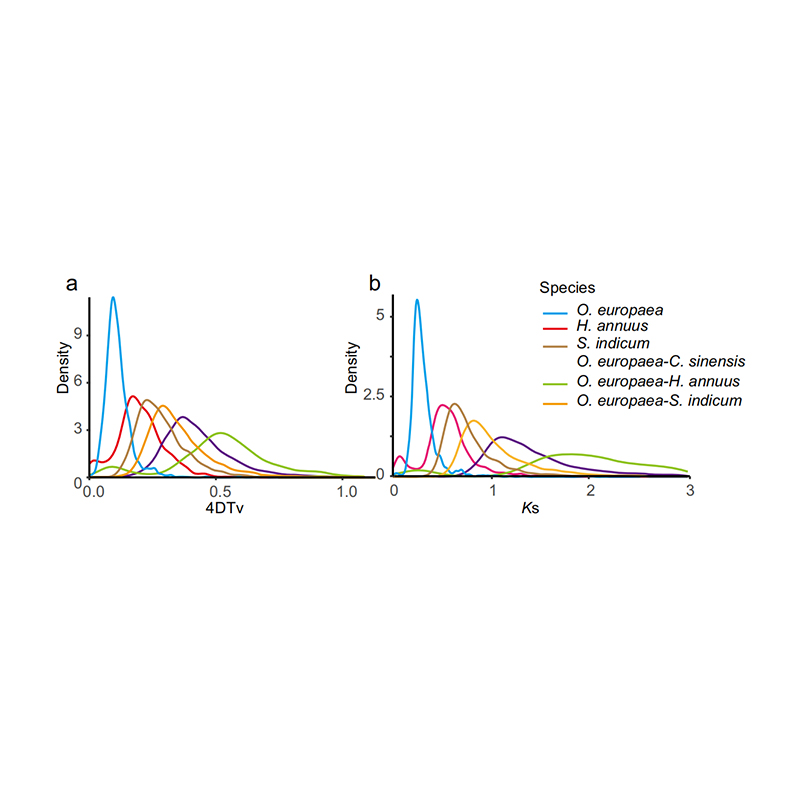
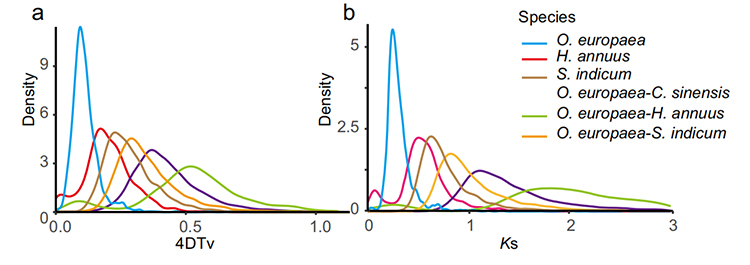
5.Análisis de duplicación del genoma completo: el análisis de distribución 4DTV y Ks mostró un evento de duplicación del genoma completo. Los picos de intraespecies mostraron eventos de duplicación. Los picos de interespecies mostraron eventos de especiación. El análisis indicó que, en comparación con las otras tres especies estrechamente relacionadas, O. europaea pasó por una duplicación genética a gran escala más recientemente.
Rao G et al.,Investigación en horticultura, 2021
Caso BMK
Rosa sin espinas: conocimientos genómicos vinculados a la adaptación a la humedad
Publicado: Revista Nacional de Ciencias, 2021
Estrategia de secuenciación:
'Basye'ssin espinas' (R.wichurainan) genoma:
Aprox. 93 X PacBio + aprox. 90 X Nanoporos + 267 X Illumina
Resultados clave
1. Se construyó el genoma de R.wichuraiana de alta calidad utilizando técnicas de secuenciación de lectura larga, que producen un ensamblaje de 530,07 Mb (el tamaño estimado del genoma fue de aproximadamente 525,9 Mb por citometría de flujo y 525,5 por estudio del genoma; la heterocigosidad fue de alrededor del 1,03%). La puntuación estimada de BUSCO fue del 93,9%. En comparación con "Old blush" (haploOB), la calidad y la integridad de este genoma se confirmaron mediante la precisión de base única y el índice de ensamblaje LTR (LAI = 20,03). El genoma de R.wichuraiana contiene 32.674 genes codificadores de proteínas.
2. El análisis conjunto multiómico, que consiste en genómica comparativa, transcriptómica y análisis QTL de la población genética, reveló la especiación crucial entre R. wichuraiana y Rosa chinensis. Además, es probable que la variación de la expresión de genes relacionados en QTL esté asociada con el patrón de espinas del tallo.
El análisis genómico comparativo entre Basye;s Thornless y Rosa chinensis, incluido el análisis de sintenia, el grupo de familias de genes y el análisis de expansión y contracción, reveló una gran cantidad de variaciones relacionadas con rasgos cruciales en las rosas. Es muy probable que la expansión única de la familia de genes NAC y FAR1/FRS esté asociada con la resistencia a la mancha negra.
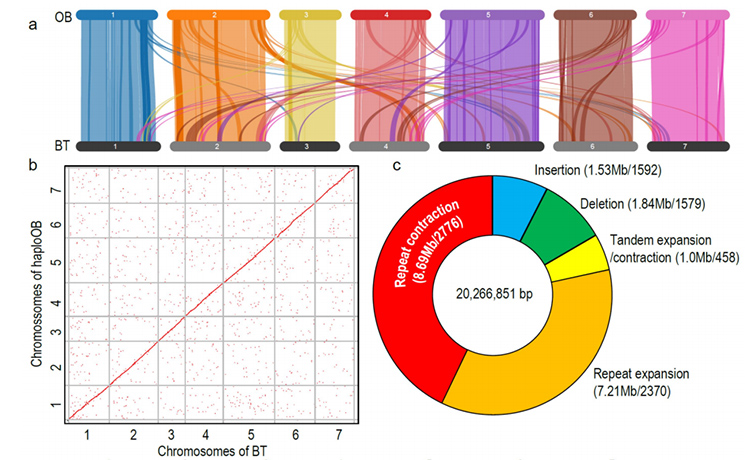

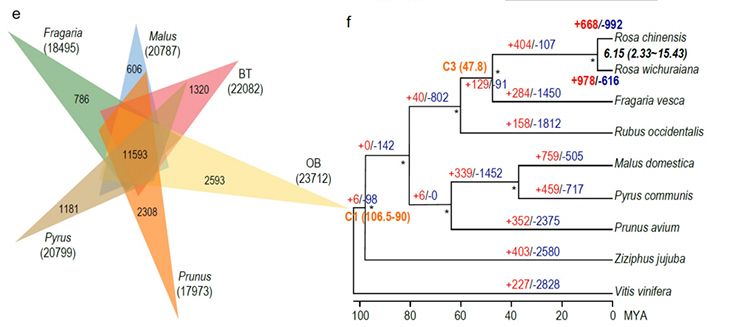
Análisis genómico comparativo entre genomas BT y haploOB.
Zhong, M., et al. “Rosa sin espinas: conocimientos genómicos vinculados a la adaptación a la humedad”Revista Nacional de Ciencias, 2021;, nwab092.





