

Análisis segregante de gran parte

Ventajas de servicio
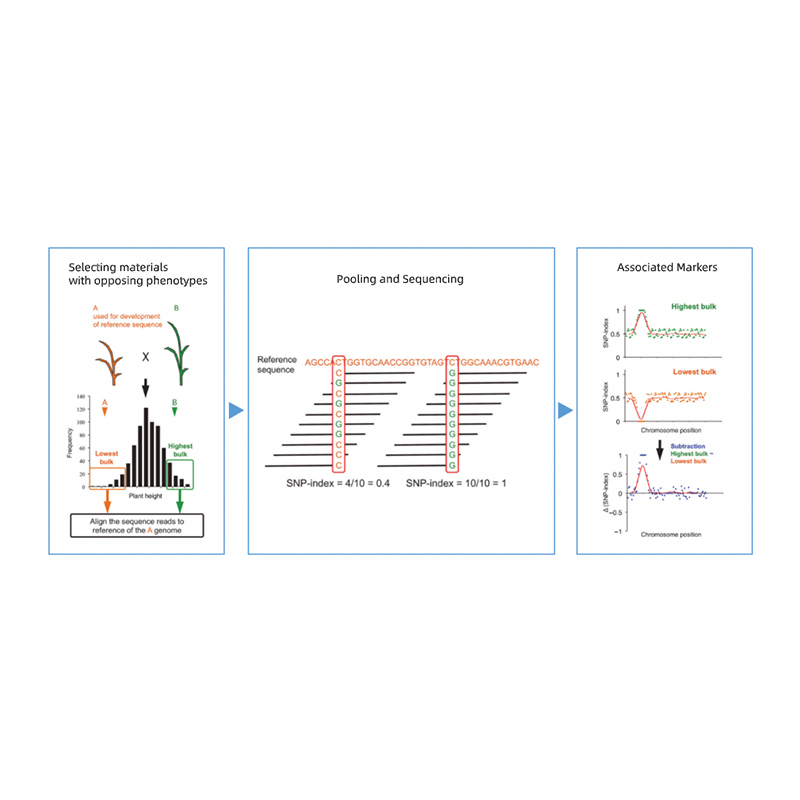
Takagi et al., The Plant Journal, 2013
● Localización precisa: mezclar a granos con 30+30 a 200+200 individuos para minimizar el ruido de fondo; Predicción de la región candidata no sinónima basada en mutatantes.
● Análisis integral: anotación de función genética candidata en profundidad, incluidas NR, SwissProt, GO, KEGG, COG, KOG, etc.
● Tiempo de respuesta más rápido: localización rápida de genes dentro de 45 días hábiles.
● Experiencia extensa: BMK ha contribuido en miles de rasgos de localización, cubriendo diversas especies como cultivos, productos acuáticos, bosques, flores, frutas, etc.
Especificaciones de servicio
Población:
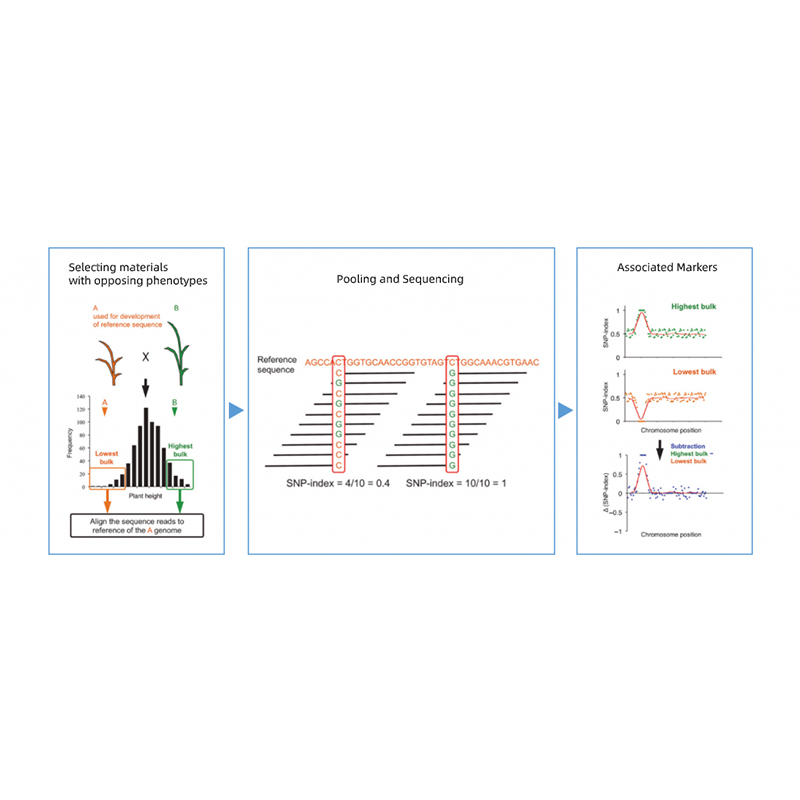
Segregación de progenie de padres con fenotipos opuestos.
p. Ej. Progenie F2, retroceso (BC), línea endogámica recombinante (RIL)
Mezcla de piscina
Para rasgos cualitativos: 30 a 50 individuos (mínimo 20)/volumen
Para Tratis cuantitativos: superiores al 5% a 10% de individuos con cualquiera de los fenotipos extremos en toda la población (mínimo 30+30).
Profundidad de secuenciación recomendada
Al menos 20x/padre y 1x/descendente
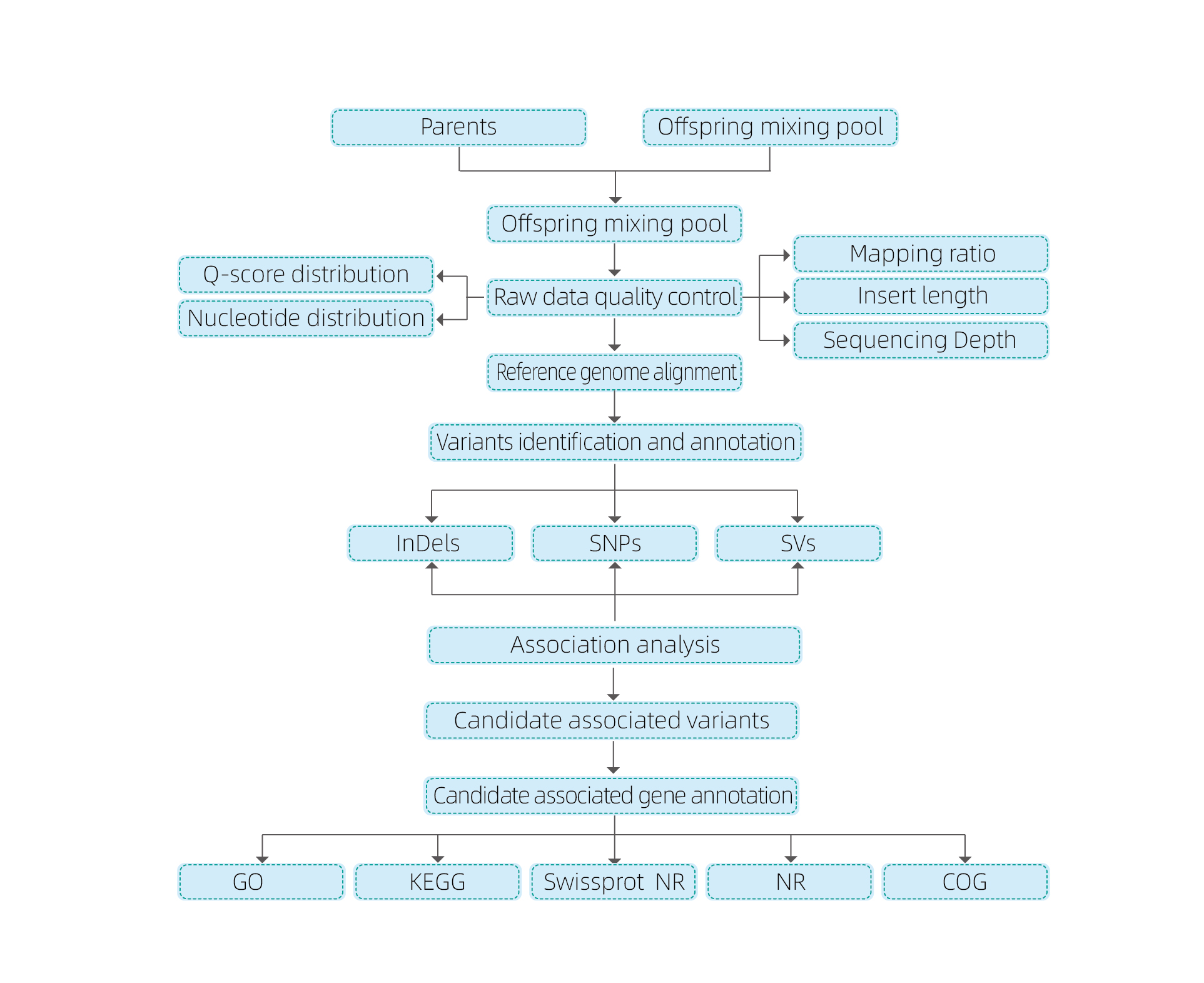
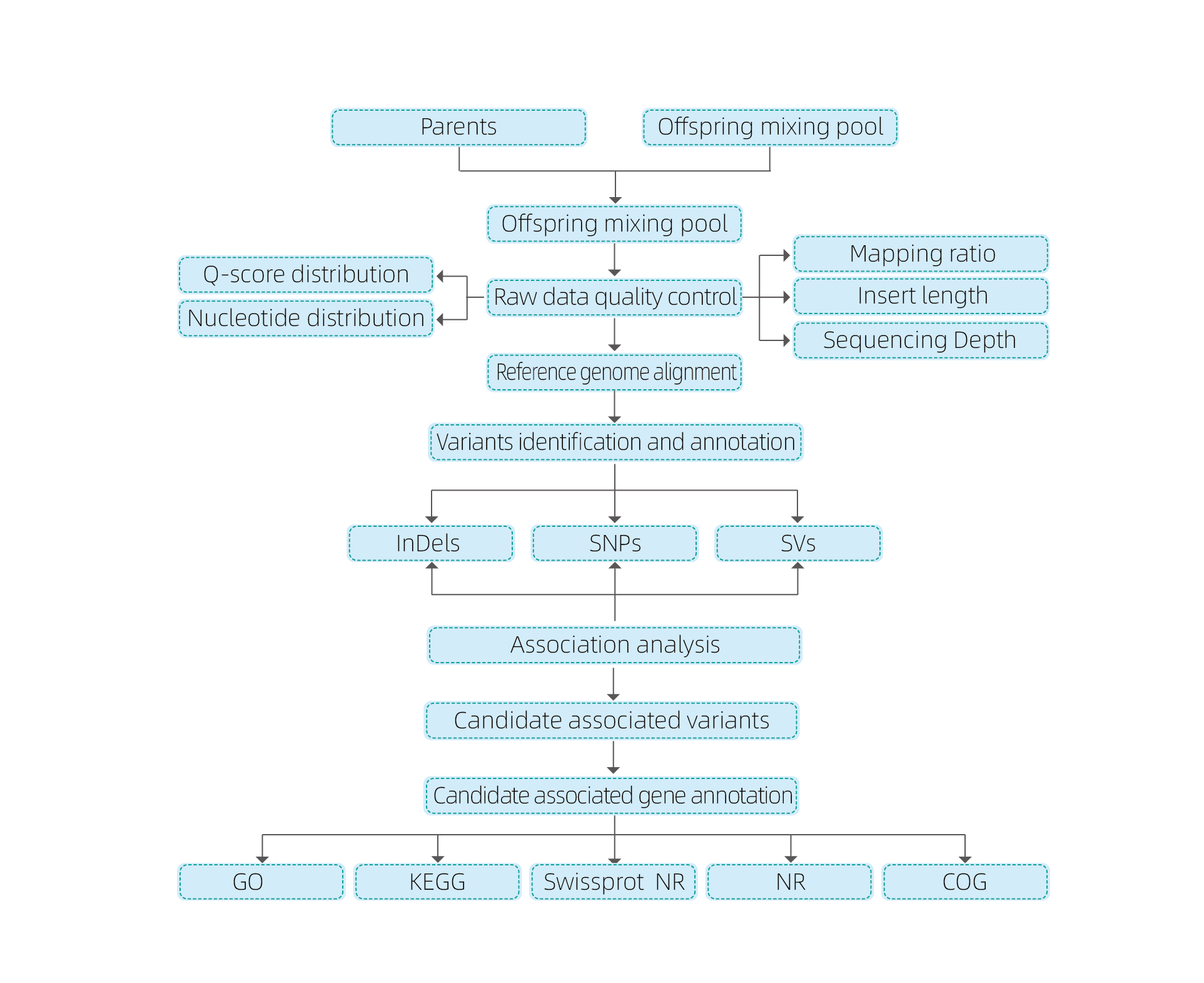
Análisis bioinformáticos
● Resequenciación del genoma completo
● Procesamiento de datos
● llamadas SNP/Indel
● Detección de la región candidata
● Anotación de la función del gen candidato
Requisitos de muestra y entrega
Requisitos de muestra:
Nucleótidos:
| muestra | Muestra de tejido |
| Concentración: ≥30 ng/μl | Plantas: 1-2 g |
| Cantidad: ≥2 μg (volumno ≥15 μl) | Animales: 0.5-1 g |
| Pureza: OD260/280 = 1.6-2.5 | Sangre entera: 1.5 ml |
Flujo de trabajo de servicio

Diseño de experimentos

Entrega de muestra

Extracción de ARN

Construcción de la biblioteca

Secuenciación

Análisis de datos

Servicios posteriores
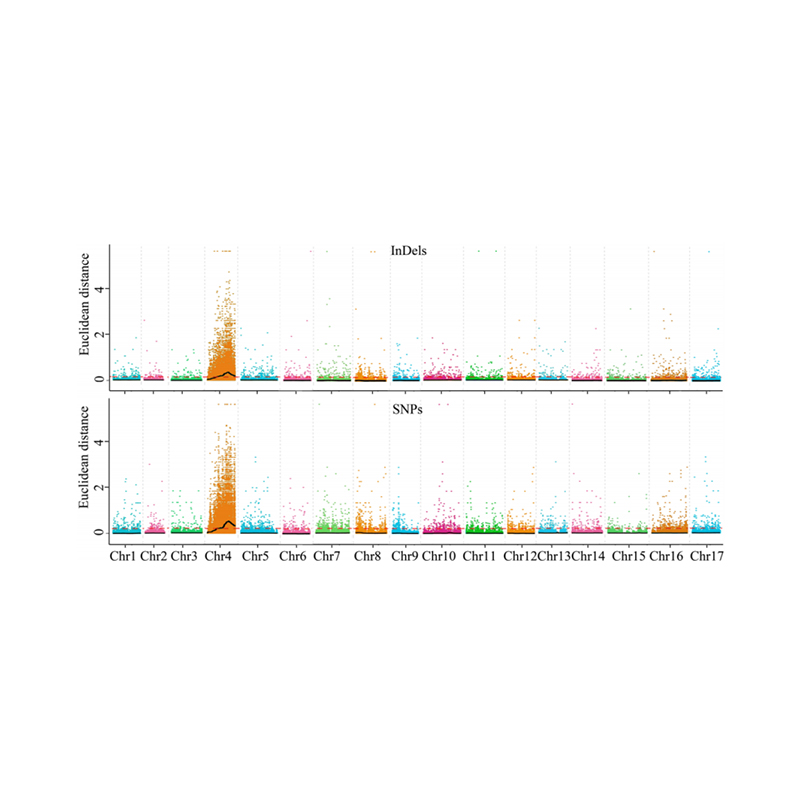
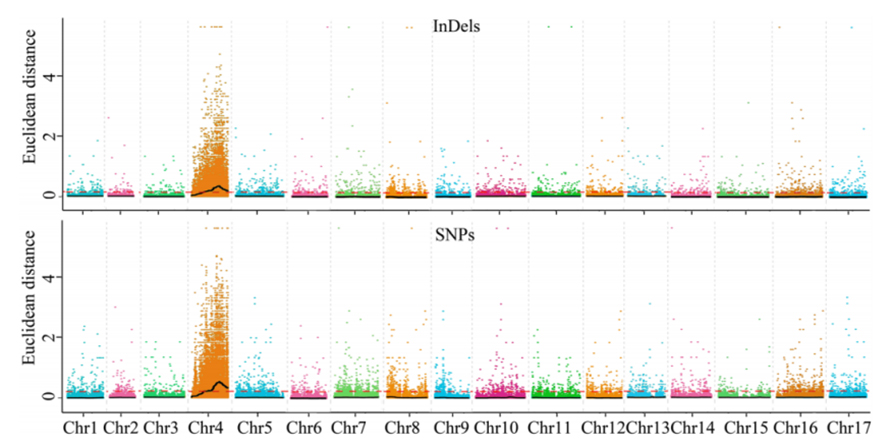
1. Base de análisis de asociación en la distancia euclidiana (ED) para identificar la región candidata. En la siguiente figura
Eje X: número de cromosoma; Cada punto representa un valor ED de un SNP. La línea negra corresponde al valor ed ajustado. Un valor de ED más alto indica una asociación más significativa entre el sitio y el fenotipo. La línea del tablero rojo representa el umbral de asociación significativa.
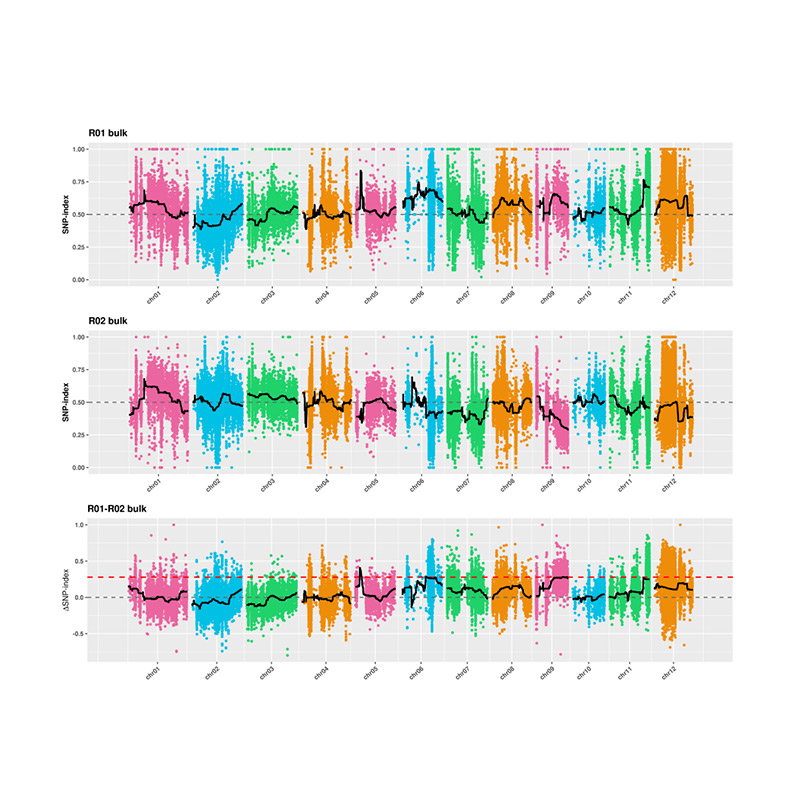
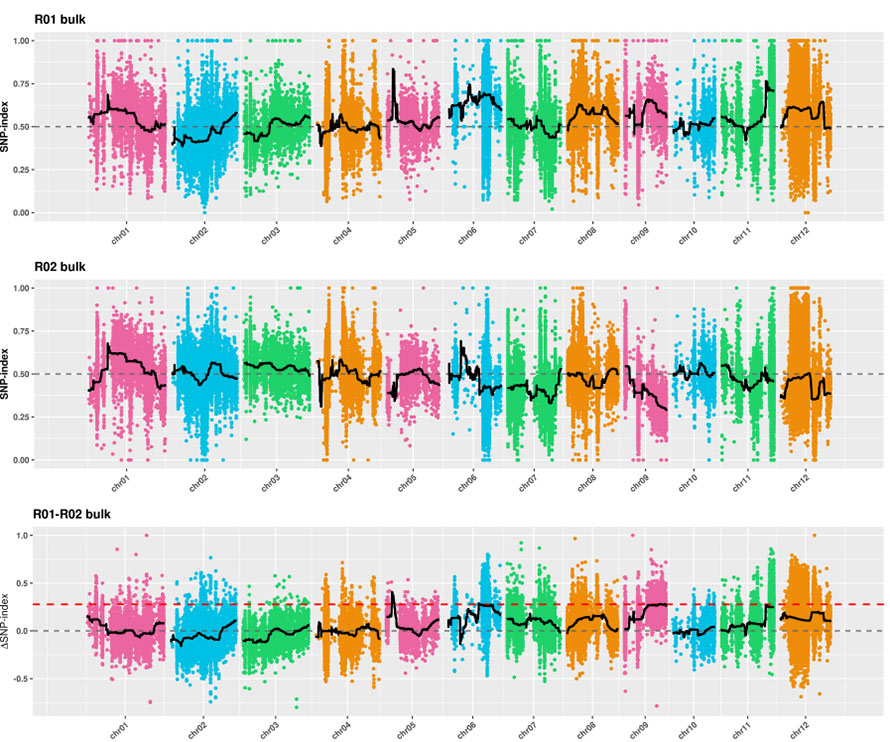
2. Análisis de asociación basado sin índice SNP
Eje X: número de cromosoma; Cada punto representa el valor de índice SNP. La línea negra significa valor de índice SNP ajustado. Cuanto mayor es el valor, más significativa es la asociación.
Caso BMK
El locus de rasgo cuantitativo de efecto mayor FNL7.1 codifica una embriogénesis tardía abundante asociada con la longitud del cuello de la fruta en el pepino
Publicado: Revista de biotecnología vegetal, 2020
Estrategia de secuenciación:
Padres (Jin5-508, YN): resequenciación del genoma completo para 34 × y 20 ×.
Piscinas de ADN (50 cuello largo y 50 cuello corto): resequenciación para 61 × y 52 ×
Resultados clave
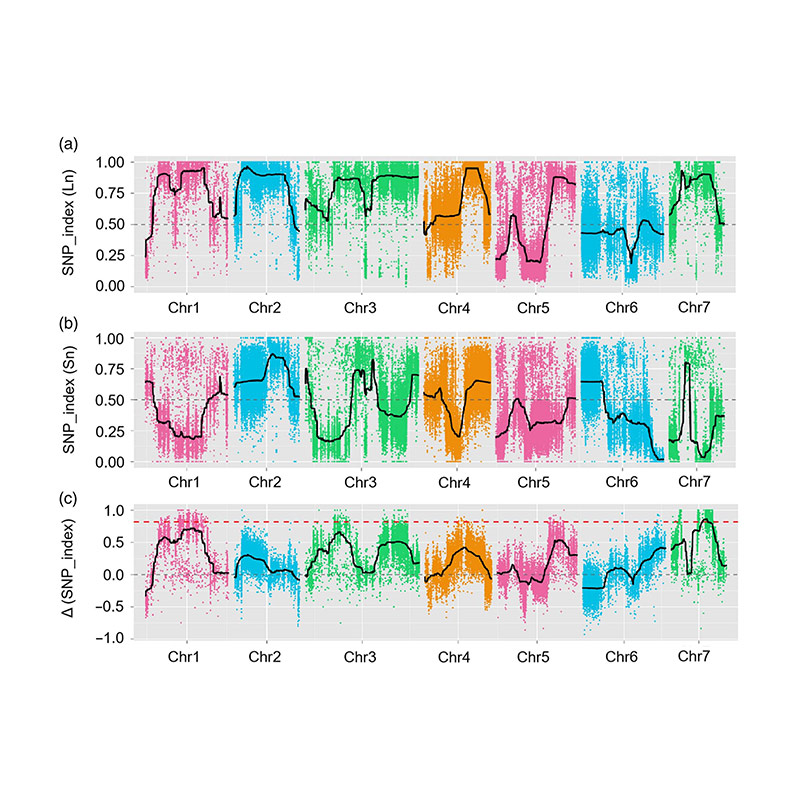
En este estudio, se generó la población segregante (F2 y F2: 3) cruzando la línea de pepino de cuello largo Jin5-508 y YN de cuello corto. Dos piscinas de ADN fueron construidas por 50 individuos extremos de cuello largo y 50 individuos extremos de cuello corto. Se identificó QTL de efecto mayor en CHR07 mediante análisis BSA y mapeo QTL tradicional. La región candidata se redujo aún más mediante mapeo fino, cuantificación de expresión génica y experimentos transgénicos, lo que reveló el gen clave en el control del cuello, CSFNL7.1. Además, se encontró que el polimorfismo en la región promotora CSFNL7.1 estaba asociada con la expresión correspondiente. El análisis filogenético adicional sugirió que es muy probable que el locus FNL7.1 se produzca de la India.
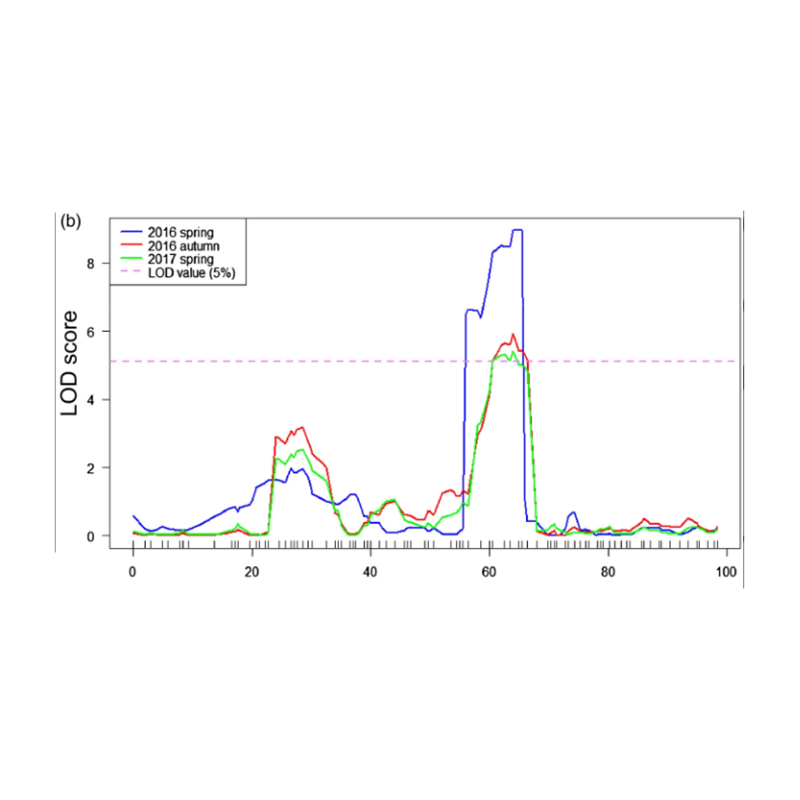
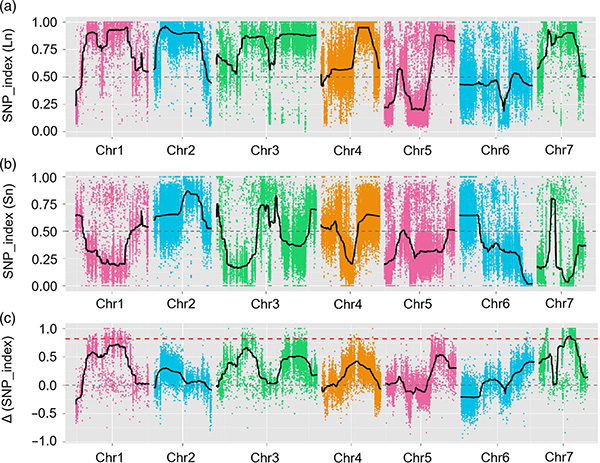 Mapeo QTL en análisis BSA para identificar la región candidata asociada con la longitud del cuello de pepino | 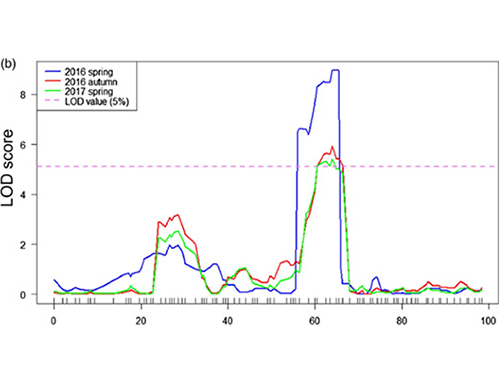 Perfiles LOD de QTL de longitud del cuello de pepino identificados en CHR07 |
Xu, X., et al. "El locus de rasgo cuantitativo de efectos mayores FNL7.1 codifica una proteína de embriogénesis tardía abundante asociada con la longitud del cuello de la fruta en el pepino". Plant Biotechnology Journal 18.7 (2020).



