

Verstärkte Fragmentsequenzierung spezifischer Lokus (SLAF-Seq)

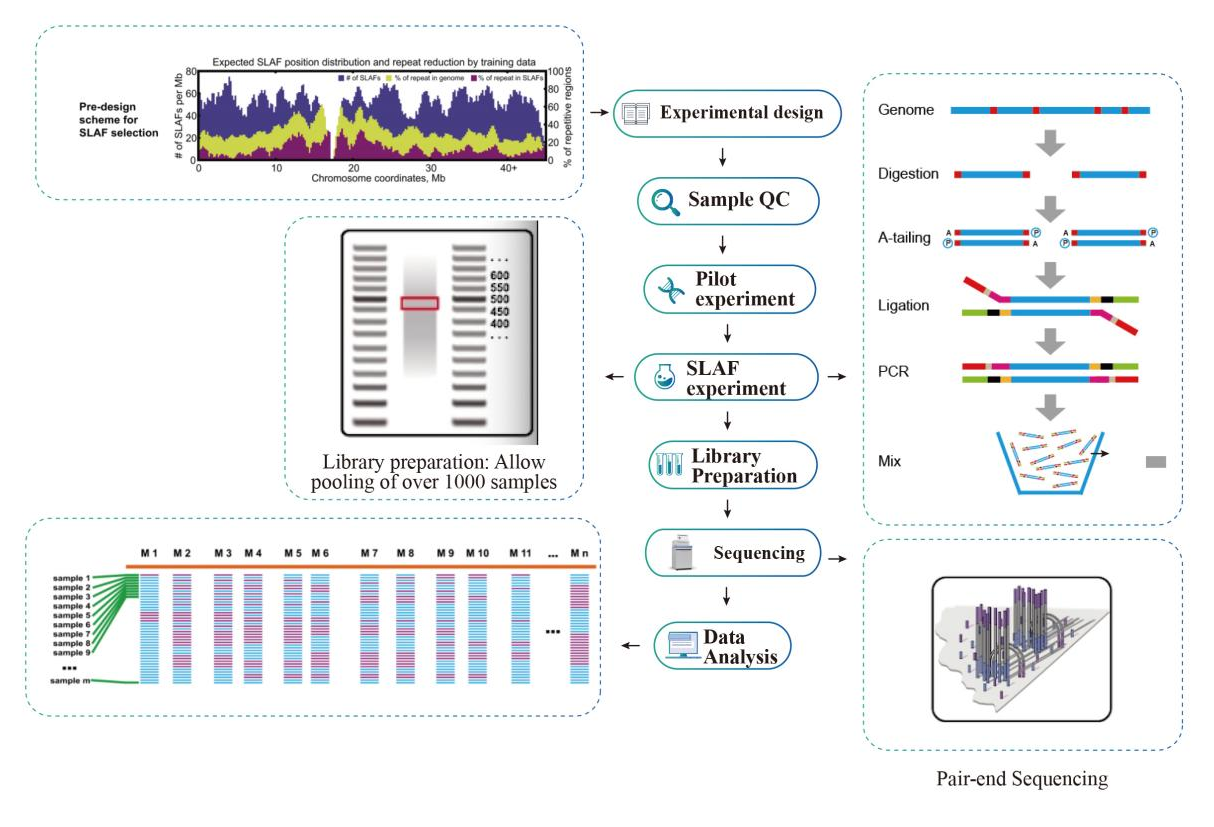
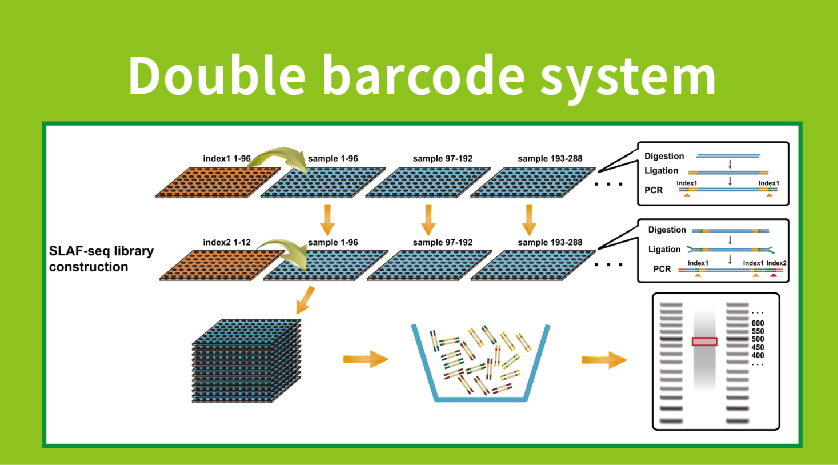
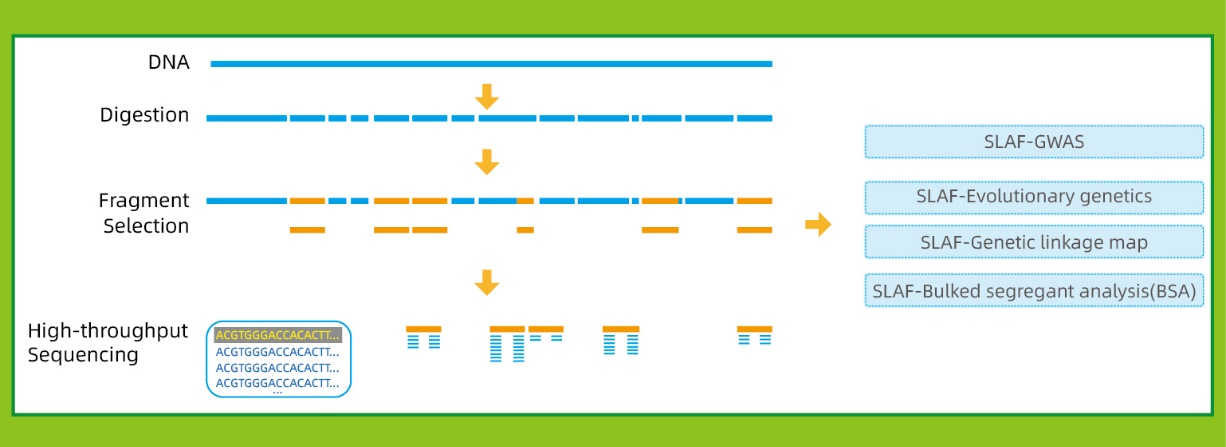
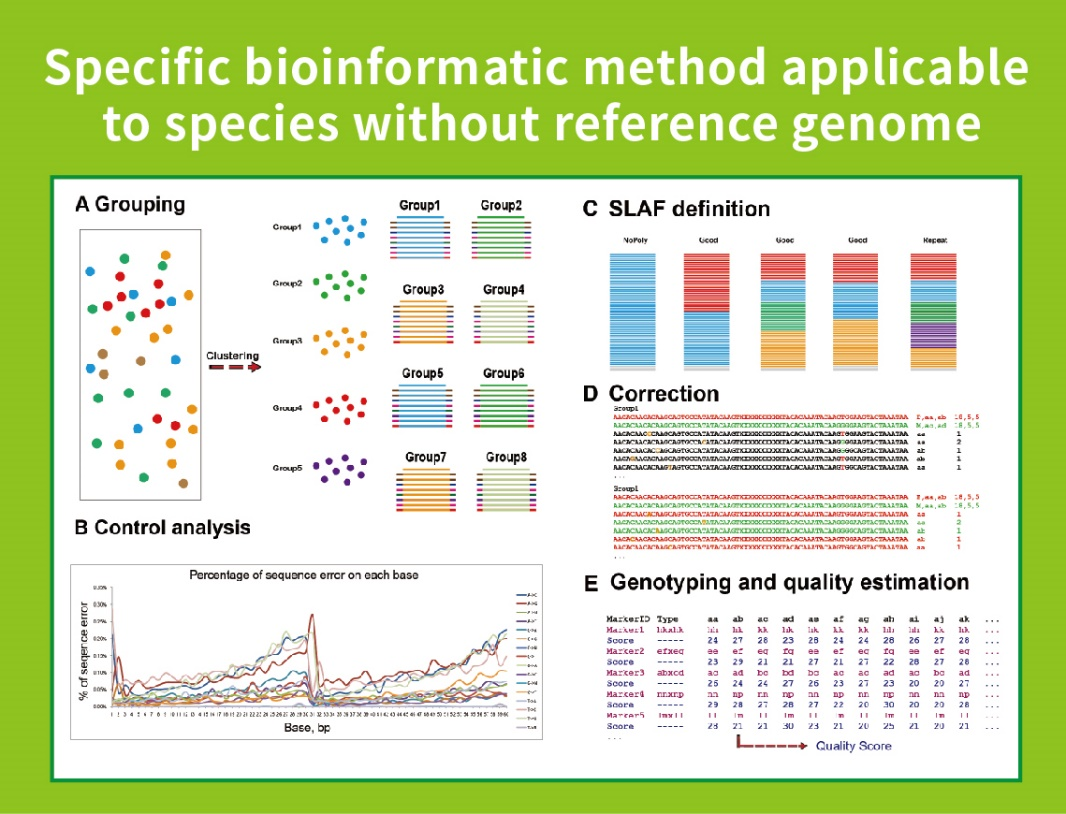
Workflow

Technisches Programm

Servicefunktionen
● Sequenzierung auf Novaseq mit PE150.
● Bibliotheksvorbereitung mit doppelter Barcodierung und Pooling von über 1000 Proben.
● Diese Technik kann mit oder ohne Referenzgenom mit unterschiedlichen bioinformatischen Pipelines für jeden Fall verwendet werden:
Mit Referenzgenom: SNP und Indel Discovery
Ohne Referenzgenom: Probenclustering und SNP -Entdeckung
● in derin SilicoVordesign Stadium Mehrfachrestriktionsenzym-Kombinationen werden untersucht, um diejenigen zu finden, die eine einheitliche Verteilung von SLAF-Tags entlang des Genoms erzeugen.
● Während des Voraussage werden drei Enzymkombinationen in 3 Proben getestet, um 9 SLAF-Bibliotheken zu generieren, und diese Informationen werden verwendet, um die optimale Kombination zur Beschränkungsenzym für das Projekt auszuwählen.
Servicevorteile
●Entdeckung mit hoher genetischer Marker: Die Integration eines Doppel-Barcode-Systems mit hohem Durchsatz ermöglicht die gleichzeitige Sequenzierung großer Populationen, und die lokusspezifische Amplifikation verbessert die Effizienz und stellt sicher, dass die Tag-Zahlen die unterschiedlichen Anforderungen verschiedener Forschungsfragen erfüllen.
● Geringe Abhängigkeit vom Genom: Es kann auf Arten mit oder ohne Referenzgenom angewendet werden.
●Flexible Schema -Design: Single-Enzym, Dual-Enzym, Multi-Enzym-Verdau und verschiedene Arten von Enzymen können alle ausgewählt werden, um unterschiedliche Forschungsziele oder -arten zu erfüllen. Derin SilicoEs wird vor dem Entwurf durchgeführt, um ein optimales Enzymdesign zu gewährleisten.
● Hohe Effizienz der enzymatischen Verdauung: Die Leitung einesin SilicoVordesign und ein vor-experiment versicherten optimales Design mit gleichmäßiger Verteilung der SLAF-Tags am Chromosom (1 SLAF-Tag/4KB) und reduzierter sich wiederholender Sequenz (<5%).
●Umfangreiches Fachwissen: Unser Team bringt eine Fülle von Erfahrungen in jedes Projekt mit sich, wobei über 5000 SLAF-Seq-Projekte an Hunderten von Arten, einschließlich Pflanzen, Säugetieren, Vögeln, Insekten und Wasserorganismen, über 5000 SLAF-Seq-Projekte geschlossen werden.
● Selbst entwickelter bioinformatischer Workflow: Bmkgene entwickelte einen integrierten bioinformatischen Workflow für SLAF-Seq, um die Zuverlässigkeit und Genauigkeit der endgültigen Ausgabe zu gewährleisten.
Servicespezifikationen
| Art der Analyse | Empfohlene Bevölkerungsskala | Sequenzierungsstrategie | |
| Tiefe der Tag -Sequenzierung | Tag -Nummer | ||
| Genetische Karten | 2 Eltern und> 150 Nachkommen | Eltern: 20x WGS OFFSPING: 10x | Genomgröße: <400 MB: WGS wird empfohlen <1 GB: 100K -Tags 1-2GB :: 200K-Tags > 2 GB: 300K -Tags Max 500K -Tags |
| Genomweite Assoziationsstudien (GWAS) | ≥200 Proben | 10x | |
| Genetische Entwicklung | ≥ 30 Proben mit> 10 Proben aus jeder Untergruppe | 10x | |
Serviceanforderungen
Konzentration ≥ 5 ng/µl
Gesamtmenge ≥ 80 ng
Nanodrop OD260/280 = 1,6-2,5
Agarosegel: Keine oder eingeschränkte Verschlechterung oder Verunreinigung
Empfohlene Probenzustellung
Behälter: 2 ml Zentrifugenröhre
(Für die meisten Proben empfehlen wir, in Ethanol nicht zu bewahren)
Beispielkennzeichnung: Proben müssen eindeutig mit dem Formular für eingereichte Beispielinformationen eingereicht und identisch sein.
Sendung: Trockeneis: Proben müssen zuerst in Taschen verpackt und in Trockeneis begraben werden.
Service Workflow






Probe QC
Pilotexperiment
Slaf-Experiment
Bibliotheksvorbereitung
Sequenzierung
Datenanalyse
Nachverkaufsdienste
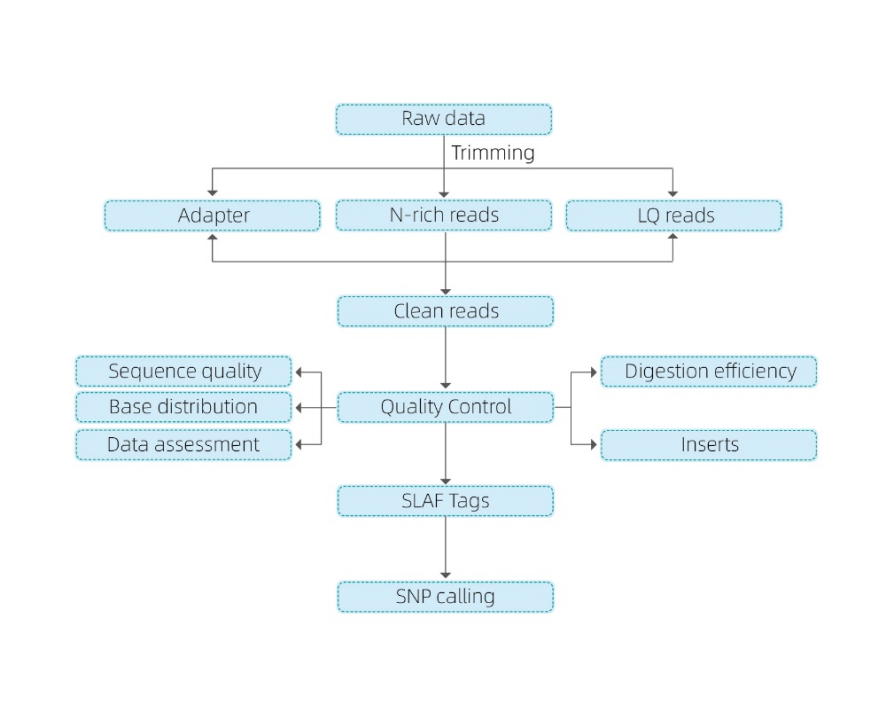 Enthält die folgende Analyse:
Enthält die folgende Analyse:

- Sequenzierungsdaten QC
- SLAF -Tagentwicklung
Mapping auf Referenzgenom
Ohne Referenzgenom: Clustering
- Analyse von SLAF -Tags: Statistik, Verteilung über das Genom
- Markierungsentdeckung: SNP, Indel, SNV, Lebenslaufanruf und Annotation
Verteilung von SLAF -Tags auf Chromosomen:

Verteilung von SNPs auf Chromosomen:
 SNP -Annotation
SNP -Annotation

| Jahr | Zeitschrift | IF | Titel | Anwendungen |
| 2022 | Naturkommunikation | 17.694 | Genomische Basis der Giga-Chromosomen und Giga-Genom von Baumpflecken Paeonia ostii | Slaf-GWAs |
| 2015 | Neuer Phytologe | 7.433 | Domestizierung Fußabdrücke verankern genomische Regionen agronomischer Bedeutung in Sojabohnen | Slaf-GWAs |
| 2022 | Journal of Advanced Research | 12.822 | Genomweite künstliche Introgressionen von Gossypium Barbadense nach G. hirsutum Entdecken Sie überlegene Loci für die gleichzeitige Verbesserung der Qualität und Ertrag von Baumwollfasern Merkmale | Slaf-Evolutionäre Genetik |
| 2019 | Molekülpflanze | 10.81 | Bevölkerungsgenomanalyse und De -novo -Assemblierung enthüllen den Ursprung von Weedy Reis als evolutionäres Spiel | Slaf-Evolutionäre Genetik |
| 2019 | Naturgenetik | 31.616 | Genomsequenz und genetische Vielfalt des gemeinsamen Karpfens Cyprinus Carpio | Slaf-Linkage-Karte |
| 2014 | Naturgenetik | 25.455 | Das Genom der kultivierten Erdnuss bietet Einblick in Hülsenfrüchte Karyotypen, polyploid Evolution und Domestizierung der Ernte. | Slaf-Linkage-Karte |
| 2022 | Plant Biotechnology Journal | 9.803 | Die Identifizierung von ST1 zeigt eine Auswahl, bei der die Samenmorphologie angehoben wird und Ölgehalt während der Soja -Domestizierung | Slaf-Marker-Entwicklung |
| 2022 | Internationales Journal of Molecular Sciences | 6.208 | Identifizierung und DNA-Markerentwicklung für einen Weizen-Leyus Mollis 2ns (2D) Disomic Chromosomensubstitution | Slaf-Marker-Entwicklung |
| Jahr | Zeitschrift | IF | Titel | Anwendungen |
| 2023 | Grenzen in der Pflanzenwissenschaft | 6.735 | QTL -Kartierung und Transkriptomanalyse des Zuckergehalts während der Fruchtreife von Pyrus pyrifolia | Genetische Karte |
| 2022 | Plant Biotechnology Journal | 8.154 | Die Identifizierung von ST1 zeigt eine Auswahl, bei der die Samenmorphologie und der Ölgehalt während der Domestizierung der Sojabohnen angehoben werden müssen
| SNP -Anruf |
| 2022 | Grenzen in der Pflanzenwissenschaft | 6.623 | Genomweite Assoziationskartierung von Hulless in der Dürreumgebung kaum Phänotypen.
| GWAS |





