

Pflanzen-/Animal de novo -Genomsequenzierung





De novoDie Sequenzierung bezieht sich auf die Konstruktion des gesamten Genoms einer Spezies unter Verwendung von Sequenzierungstechnologien ohne Referenzgenom. Die Einführung und die weit verbreitete Übernahme der Sequenzierung der dritten Generation mit längeren Lesevorgängen haben die Genomanordnung durch Erhöhen der Überlappung zwischen den Lesevorgängen signifikant verbessert. Diese Verbesserung ist besonders relevant im Umgang mit herausfordernden Genomen, wie z. allein.
Unsere One-Stop-Lösung bietet integrierte Sequenzierungsdienste und bioinformatische Analysen, die ein hochwertiges De-novo-Genom liefern. Eine erste Genomumfrage mit Illumina liefert Schätzungen der Genomgröße und -komplexität. Diese Informationen werden verwendet, um den nächsten Schritt der langlebigen Sequenzierung mit Pacbio HiFi zu leiten, gefolgt vonde novoAnsammlung von Contigs. Die anschließende Verwendung der HIC-Anordnung ermöglicht die Verankerung der Contigs an dem Genom und erhalten eine Ansammlung von Chromosomenebene. Schließlich wird das Genom durch die Genvorhersage und durch Sequenzierung exprimiertes Gene annotiert, die auf Transkriptome mit kurzen und langen Lesevorgängen zurückgreifen.
Servicefunktionen
● Integration mehrerer Sequenzierung und bioinformatischer Dienste in eine One-Stop-Lösung:
Genomumfrage mit Illumina, um die Genomgröße abzuschätzen und nachfolgende Schritte zu leiten;
Lange gelesene Sequenzierung fürde novoVersammlung von Contigs;
Hi-C-Sequenzierung für Chromosomenankereien;
mRNA -Sequenzierung zur Annotation von Genen;
Validierung der Baugruppe.
● Service, der für die konstruierenden neuen Genome oder Verbesserung bestehender Referenzgenome für Arten von Interesse geeignet ist.
Servicevorteile
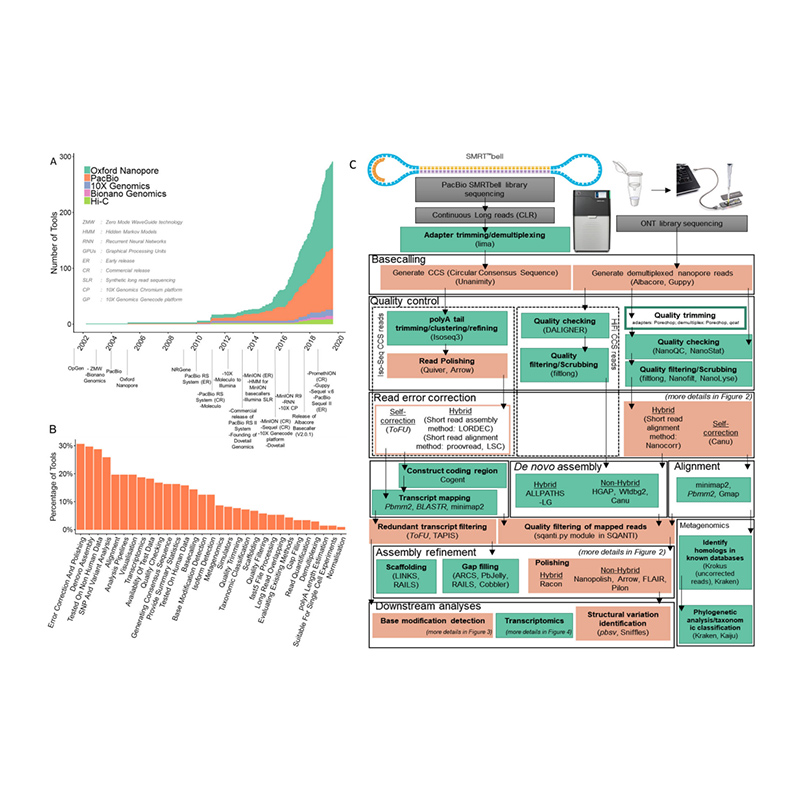
Entwicklung von Sequenzierungsplattformen und Bioinformatik inde novoGenomanordnung
(Amarasinghe SL et al.,,Genombiologie, 2020)
●Umfangreiches Fachwissen und Veröffentlichungsaufzeichnung: BMKgene hat massive Erfahrung in hochwertiger Genomanordnung verschiedener Arten angesammelt, einschließlich diploiden Genomen und hochkomplexen Genomen von polyploidischen und allpolypliden Arten. Seit 2018 haben wir zu Over beigetragen300 Hochwirkungsveröffentlichungen und 20+ von ihnen werden in Nature Genetics veröffentlicht.
● One-Stop-Lösung: Unser integrierter Ansatz kombiniert mehrere Sequenzierungstechnologien und bioinformatische Analysen zu einem zusammenhängenden Workflow und liefert ein hochwertiges zusammengestelltes Genom.
●Auf Ihre Bedürfnisse zugeschnitten: Unser Service -Workflow ist anpassbar und ermöglicht die Anpassung an Genome mit unterschiedlichen Merkmalen und spezifischen Forschungsbedürfnissen. Dazu gehören die zuvorkommenden riesigen Genome, polyploiden Genome, hoch heterozygoten Genome und mehr.
●Hochqualifiziertes Bioinformatik und Laborteam: Mit großer Erfahrung sowohl in der experimentellen als auch in der Bioinformatik -Front komplexer Genomanordnungen und einer Reihe von Patenten und Software -Urheberrechten.
●Unterstützung nach dem Verkauf:Unser Engagement geht über den Abschluss des Projekts hinaus mit einer 3-monatigen Nachverkaufsdauer hinaus. Während dieser Zeit bieten wir Projekt-Follow-up, Fehlerbehebung und Q & A-Sitzungen an, um alle Abfragen im Zusammenhang mit den Ergebnissen zu beheben.
Servicespezifikationen
| Genomumfrage | Genomanordnung | Chromosomenebene | Genomannotation |
| 50x Illumina Novaseq PE150
| 30x Pacbio CCS HiFi liest | 100x Hi-C | RNA-Seq Illumina PE150 10 GB + (optional) RNA-seq pacbio in voller Länge 40 GB oder Nanopore 12 GB |
Serviceanforderungen
Für die Genomumfrage, die Genomanordnung und die Hi-C-Assemblierung:
| Gewebe oder extrahierte Nukleinsäuren | Genomumfrage | Genomanordnung mit Pacbio | Hi-C-Baugruppe |
| Tierviscera | 0,5-1 g
| ≥ 3,5 g | ≥2 g |
| Tiermuskel | ≥ 5 g | ||
| Blutes Säugetier | 1,5 ml
| ≥ 5 ml | ≥2 ml |
| Geflügel/Fischblut | ≥ 0,5 ml | ||
| Pflanzlich- frisches Blatt | 1-2 g | ≥ 5 g | ≥ 4 g |
| Kultivierte Zellen |
| ≥ 1x108 | ≥ 1x107 |
| Insekt | 0,5-1 g | ≥ 3 g | ≥ 2 g |
| Extrahierte DNA | Konzentration: ≥ 1 ng/ µl Menge ≥ 30 ng Begrenzte oder keine Verschlechterung oder Verschmutzung | Konzentration: ≥ 50 ng/ µl Menge: 10 µg/Durchflusszelle/Probe OD260/280 = 1,7-2,2 OD260/230 = 1,8-2,5 Begrenzte oder keine Verschlechterung oder Verschmutzung |
-
|
Für Genomanmerkungen mit Transkriptomik:
| Gewebe oder extrahierte Nukleinsäuren | Illumina -Transkriptom | Pacbio -Transkriptom | Nanopore -Transkriptom |
| Pflanzenwurzel/Stamm/Blütenblatt | 450 mg | 600 mg | |
| Pflanze - Blatt/Samen | 300 mg | 300 mg | |
| Pflanze - Obst | 1,2 g | 1,2 g | |
| Tierherz/Darm | 300 mg | 300 mg | |
| Tierviscera/Gehirn | 240 mg | 240 mg | |
| Tiermuskel | 450 mg | 450 mg | |
| Tierknochen/Haar/Haut | 1 g | 1 g | |
| Arthropod - Insekt | 6 | 6 | |
| Arthropod -Crustacea | 300 mg | 300 mg | |
| Vollblut | 1 Rohr | 1 Rohr | |
| Extrahierte RNA | Konzentration: ≥ 20 ng/ µl Menge ≥ 0,3 µg OD260/280 = 1,7-2,5 OD260/230 = 0,5-2,5 Rin≥ 6 5 ≥ 28S/18S ≥ 1 | Konzentration: ≥ 100 ng/ µl Menge ≥ 0,75 µg OD260/280 = 1,7-2,5 OD260/230 = 0,5-2,5 Rin≥ 8 5 ≥ 28S/18S ≥ 1 | Konzentration: ≥ 100 ng/ µl Menge ≥ 0,75 µg OD260/280 = 1,7-2,5 OD260/230 = 0,5-2,5 Rin≥ 7,5 5 ≥ 28S/18S ≥ 1 |
Empfohlene Probenzustellung
Behälter: 2 ml Zentrifugenrohr (Zinnfolie wird nicht empfohlen)
(Für die meisten Proben empfehlen wir, in Ethanol nicht zu bewahren.)
Beispielkennzeichnung: Proben müssen klar gekennzeichnet und identisch mit dem eingereichten Beispielinformationsformular markiert und identisch sein.
Sendung: Trockeneis: Proben müssen zuerst in Taschen verpackt und in Trockeneis begraben werden.
Workflow

Service Work Flow

Experimententwurf

Probenabgabe

DNA -Extraktion

Bibliothekskonstruktion

Sequenzierung

Datenanalyse

Nachverkaufsdienste

Komplette bioinformatische Analyse, getrennt in 4 Schritten:
1) Genome Survey basierend auf der K-MER-Analyse mit NGS liest:
Schätzung der Genomgröße
Schätzung der Heterozygotie
Schätzung der sich wiederholenden Regionen
2) Genomanordnung mit Pacbio HiFi:
De novoMontage
Assembly Assessment: einschließlich Busco -Analyse für die Vollständigkeit und die Kartierung von NGS und Pacbio HiFi Reads
3) Hi-C-Baugruppe:
HI-C-Bibliothek QC: Schätzung gültiger Hi-C-Interaktionen
Hi-C-Assemblierung: Clustering von Contigs in Gruppen, gefolgt von der ContIG-Bestellung in jeder Gruppe und zugewiesener Contig-Orientierung
Hi-C-Bewertung
4) Genomannotation:
Nichtkodierende RNA-Vorhersage
Repetitive Sequenzen Identifikation (Transposons und Tandem -Wiederholungen)
Genvorhersage
§De novo: ab Initio Algorithmen
§ basierend auf Homologie
§ basierend auf Transkriptom, mit langen und kurzen Lesevorgängen: Lesevorgänge sindde novozusammengebaut oder dem Entwurfsgenom zugeordnet
§ Annotation von vorhergesagten Genen mit mehreren Datenbanken
1) Genome Survey-K-MER-Analyse

2) Genomanordnung

2) Genom -Assemblierung - Pacbio HiFi liest Mapping to Draft Assembly

2) HI-C-Assemblierung-Schätzung von Hi-C-gültigen Interaktionspaaren

3) Hi-C-Bewertung nach der Assembly

4) Genomannotation - Integration vorhergesagter Gene

4) Genomannotation - vorhergesagte Geneannotation

Erforschen Sie die Fortschritte, die von BMKgene De novo Genom Assembly Services durch eine kuratierte Sammlung von Veröffentlichungen erleichtert wurden:
Li, C. et al. (2021) "Genomsequenzen zeigen globale Ausbreitungsrouten und deuten auf konvergente genetische Anpassungen in der Seahorse -Evolution hin“, Nature Communications, 12 (1). doi: 10.1038/s41467-021-21379-x.
Li, Y. et al. (2023) 'groß angelegte chromosomale Veränderungen führen zu Veränderungen auf Genomebene, Umweltanpassung und Speziation im Gayal (BOS-Frontalis), Molekularbiologie und Evolution, 40 (1). doi: 10.1093/molbev/msad006.
Tian, T. et al. (2023) "Genomansammlung und genetische Dissektion eines prominenten dürreresistenten Maiskeimplasmas", Naturgenetik 2023 55: 3, 55 (3), S. 496–506. doi: 10.1038/s41588-023-01297-y.
F. et al. (2023) „Aufschluss über die Evolution der Tropane -Alkaloid -Biosynthese durch Analyse von zwei Genomen in der Solanaceae -Familie“, Nature Communications 2023 14: 1, 14 (1), S. 1–18. doi: 10.1038/s41467-023-37133-4.
Herausfordernde Fallstudien:
Telomer-zu-Telomer-Versammlung:Fu, A. et al. (2023) "Telomer-Telomer-Genom-Assemblierung von bitterer Melone (Momordica Charantia L. Var. Abbreviata Ser.) Zeigt die genetische Merkmale der Fruchtentwicklung, Zusammensetzung und Reifung", Horticulture Research, 10 (1). doi: 10.1093/hr/UHAC228.
Haplotyp -Baugruppe:Hu, W. et al. (2021) "Alleldefiniertes Genom zeigt eine biallelische Differenzierung während der Maniokentwicklung", molekulare Pflanze, 14 (6), S. 851–854. doi: 10.1016/j.molp.2021.04.009.
Riesengenomanordnung:Yuan, J. et al. (2022) 'Genomische Grundlage der Giga-Chromosomen und des Giga-Genoms von Baumpfähen Paeonia ostii', Nature Communications 2022 13: 1, 13 (1), S. 1–16. doi: 10.1038/s41467-022-35063-1.
Polyploide Genomanordnung:Zhang, Q. et al. (2022) "Genomische Einblicke in die jüngste Chromosomenreduktion von autopolyploidem Zuckerrohr Saccharum spontaneum", Nature Genetics 2022 54: 6, 54 (6), S. 885–896. doi: 10.1038/s41588-022-01084-1.





