

Evolutionäre Genetik

Servicevorteile
Takagi et al.,Das Pflanzenjournal, 2013
●Umfassende bioinformatische Analyse:Ermöglicht die Abschätzung der genetischen Vielfalt, die das evolutionäre Potenzial von Arten widerspiegelt, und zeigt zuverlässige phylogenetische Beziehungen zwischen Arten auf, wobei der Einfluss konvergenter und paralleler Evolution minimiert wird
●Optionale kundenspezifische Analyse: wie die Schätzung der Divergenzzeit und -geschwindigkeit basierend auf Variationen auf Nukleotid- und Aminosäureebene.
●Umfangreiche Expertise und Publikationsaufzeichnungen: BMKGene hat über 15 Jahre lang umfangreiche Erfahrung in Projekten zur Populations- und Evolutionsgenetik gesammelt, die Tausende von Arten usw. abdecken, und zu über 1000 hochrangigen Projekten beigetragen, die in Nature Communications, Molecular Plants, Plant Biotechnology Journal usw. veröffentlicht wurden.
● Hochqualifiziertes Bioinformatik-Team und kurze Analysezyklen: Mit großer Erfahrung in der fortgeschrittenen Genomanalyse liefert das Team von BMKGene umfassende Analysen mit kurzer Bearbeitungszeit.
● Post-Sales-Support:Unser Engagement geht über den Projektabschluss hinaus und umfasst einen dreimonatigen Kundendienstzeitraum. Während dieser Zeit bieten wir Projektnachverfolgung, Unterstützung bei der Fehlerbehebung und Frage-und-Antwort-Sitzungen an, um alle Fragen zu den Ergebnissen zu beantworten.
Leistungsbeschreibung und Anforderungen
| Art der Sequenzierung | Empfohlene Bevölkerungsskala | Sequenzierungsstrategie | Nukleotidanforderungen |
| Sequenzierung des gesamten Genoms | ≥ 30 Personen, mit ≥ 10 Personen aus jeder Untergruppe
| 10x | Konzentration: ≥ 1 ng/µL Gesamtmenge ≥ 30 ng Begrenzte oder keine Verschlechterung oder Kontamination |
| Specific-Locus Amplified Fragment (SLAF) | Tag-Tiefe: 10x Anzahl der Tags: <400 MB: WGS wird empfohlen <1 GB: 100.000 Tags 1 GB >2 GB: 300.000 Tags Maximal 500.000 Tags | Konzentration ≥ 5 ng/µL Gesamtmenge ≥ 80 ng Nanodrop OD260/280=1,6-2,5 Agarosegel: kein oder begrenzter Abbau oder Kontamination
|
Service-Workflow

Experimentdesign

Musterlieferung

Bibliotheksbau

Sequenzierung

Datenanalyse

Kundendienst

Der Service umfasst die Analyse der Populationsstruktur (phylogenetischer Baum, PCA, Populationsschichtungsdiagramm), der Populationsvielfalt und der Populationsauswahl (Verknüpfungsungleichgewicht, selektive Sweep-Auswahl vorteilhafter Standorte). Der Service kann auch kundenspezifische Analysen umfassen (z. B. Divergenzzeit, Genfluss).
*Die hier gezeigten Demo-Ergebnisse stammen alle von Genomen, die mit BMKGENE veröffentlicht wurden
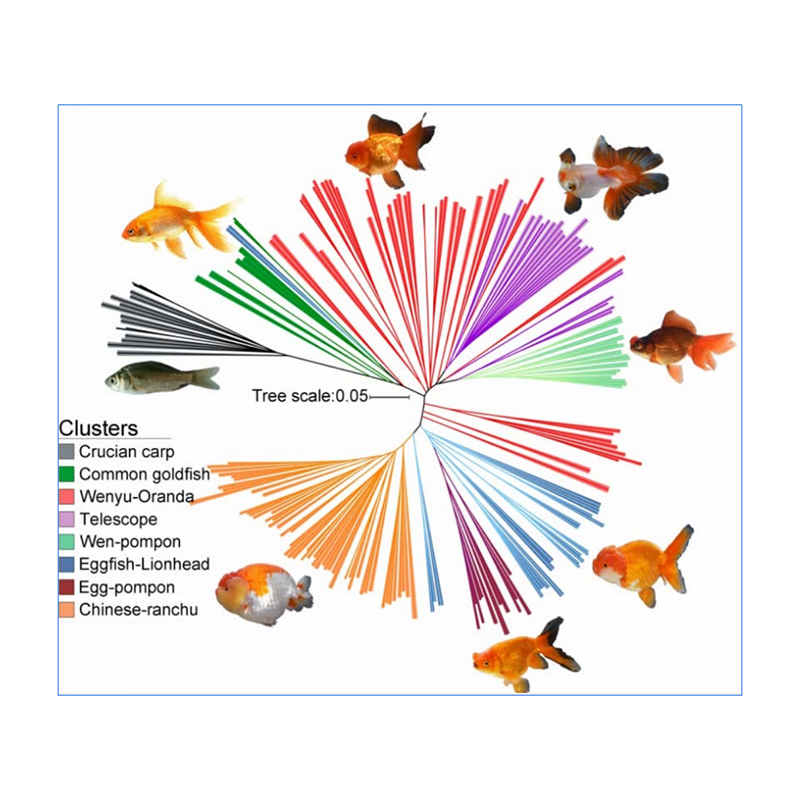
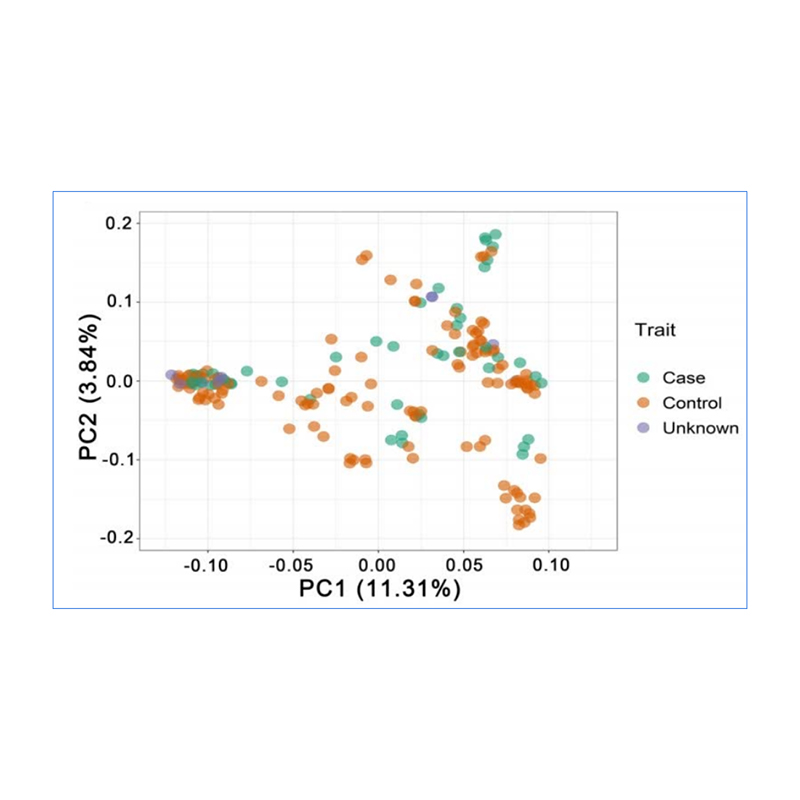
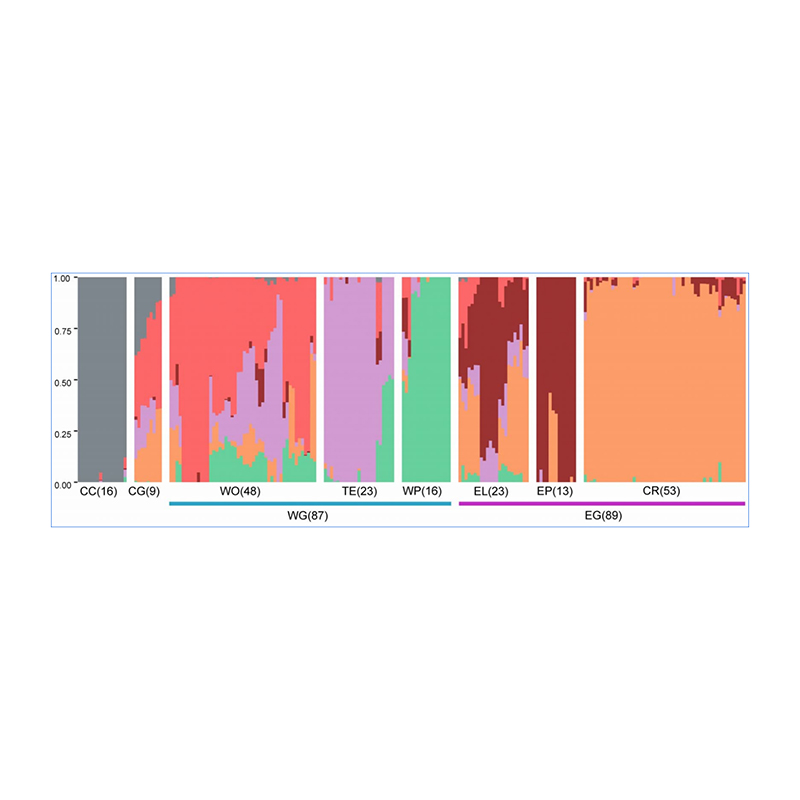
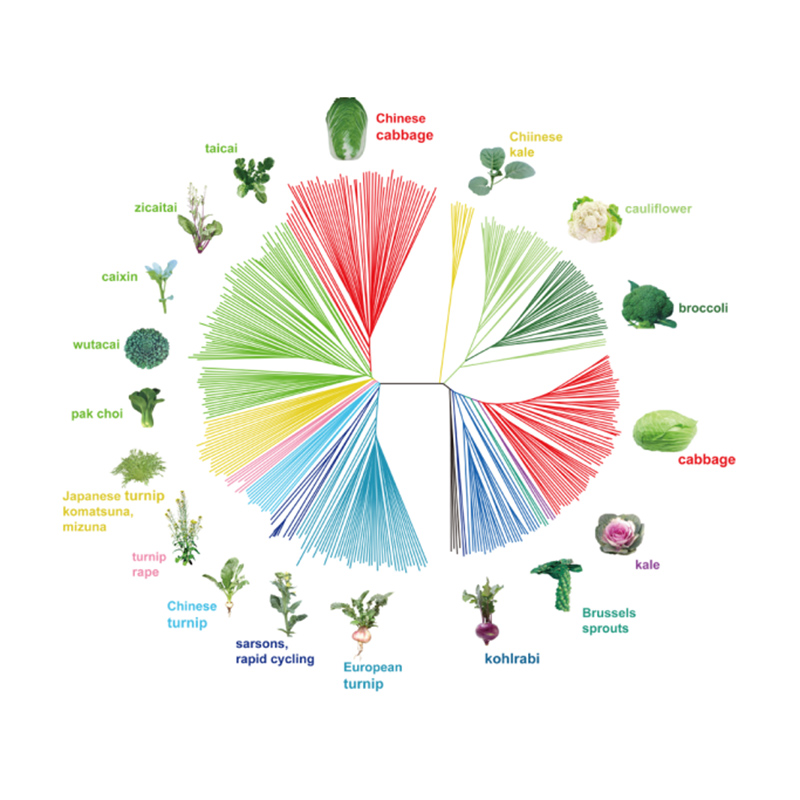
1. Die Evolutionsanalyse umfasst die Konstruktion eines Stammbaums, einer Populationsstruktur und einer PCA auf der Grundlage genetischer Variationen.
Der phylogenetische Baum stellt taxonomische und evolutionäre Beziehungen zwischen Arten mit gemeinsamen Vorfahren dar.
PCA zielt darauf ab, die Nähe zwischen Teilpopulationen zu visualisieren.
Die Populationsstruktur zeigt das Vorhandensein einer genetisch unterschiedlichen Subpopulation hinsichtlich der Allelhäufigkeiten.
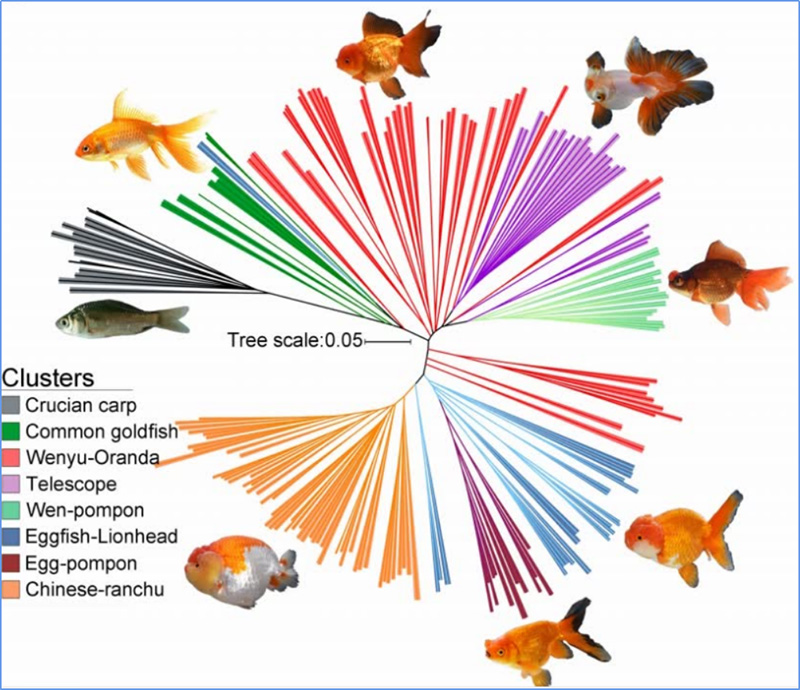
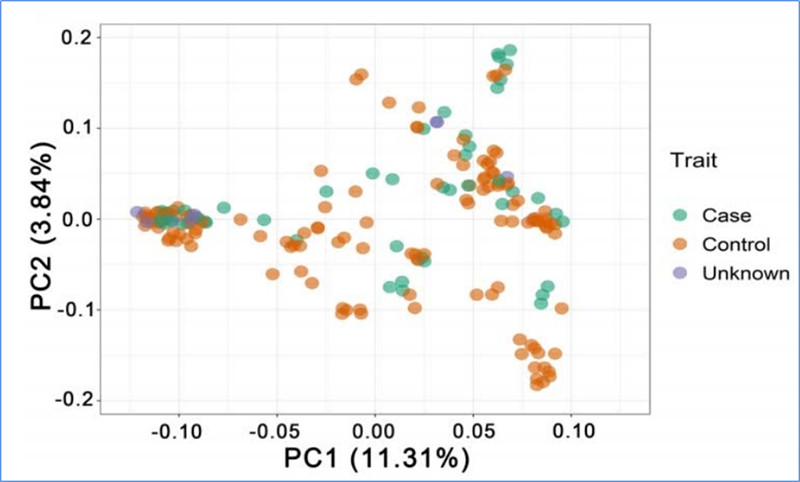
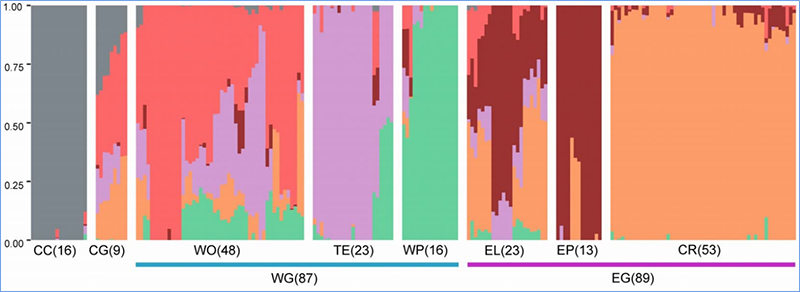
Chen et al. al.,PNAS, 2020
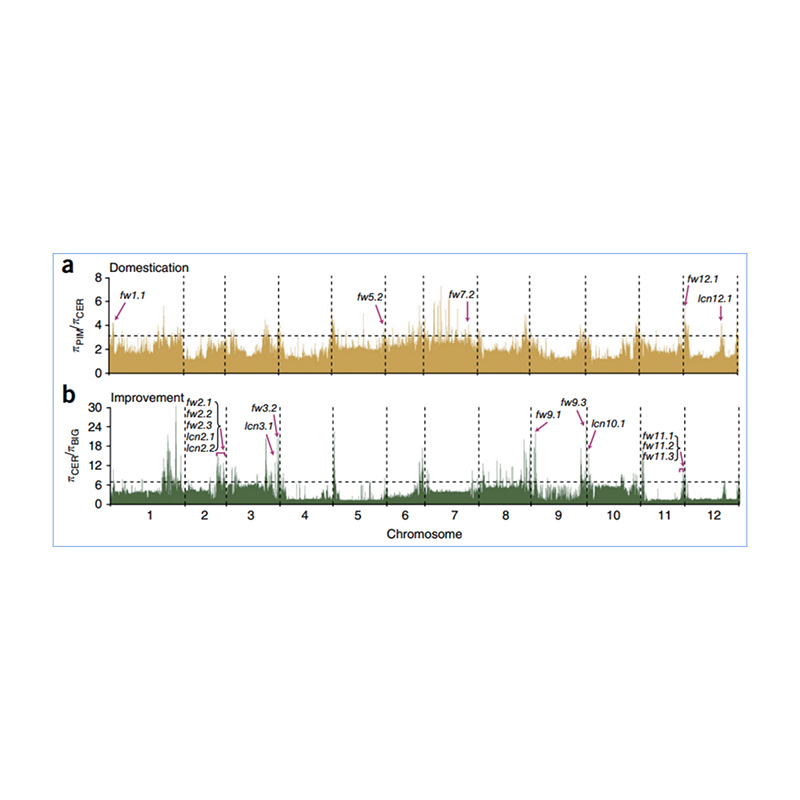
2. Selektiver Sweep
Unter selektivem Sweep versteht man einen Prozess, bei dem ein vorteilhafter Standort ausgewählt wird und die Häufigkeit verknüpfter neutraler Standorte erhöht und die Häufigkeit nicht verknüpfter Standorte verringert wird, was zu einer Verringerung regionaler Standorte führt.
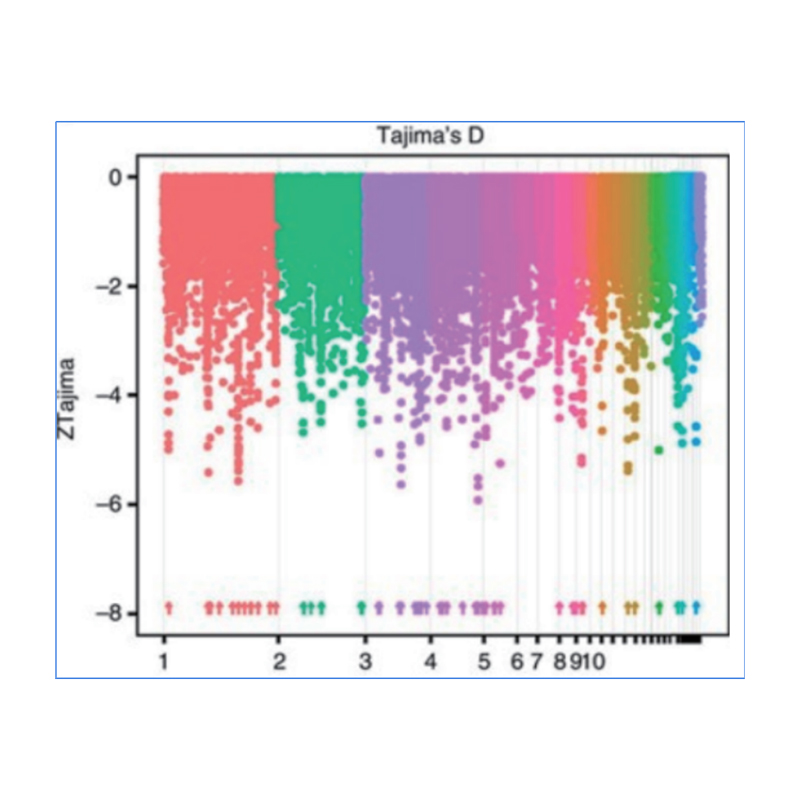
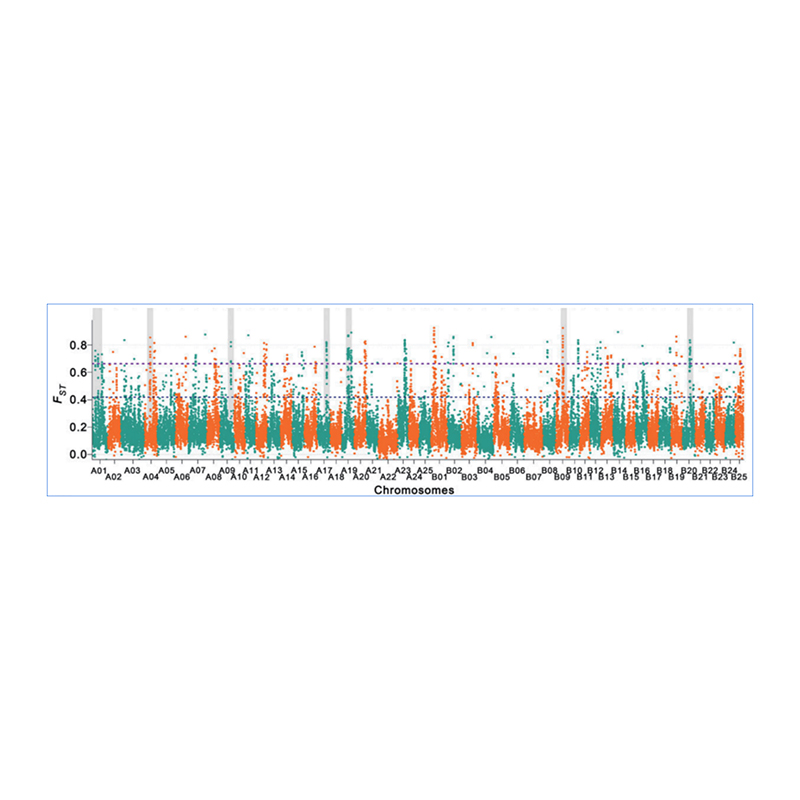
Der genomweite Nachweis in selektiven Sweep-Regionen erfolgt durch Berechnung des populationsgenetischen Index (π,Fst, Tajima's D) aller SNPs innerhalb eines Schiebefensters (100 Kb) in einem bestimmten Schritt (10 Kb).
Nukleotidvielfalt(π)
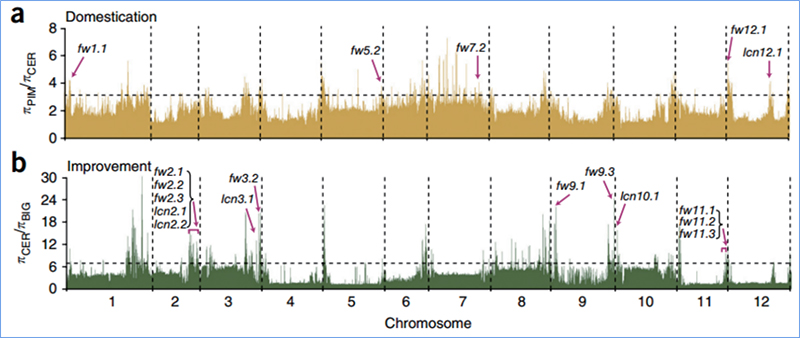
Tajimas D
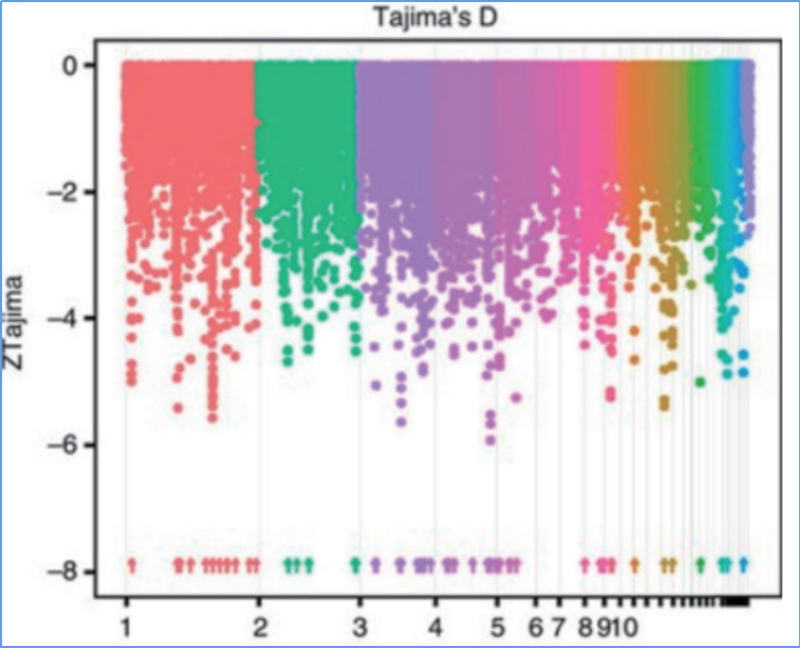
Fixationsindex (Fst)
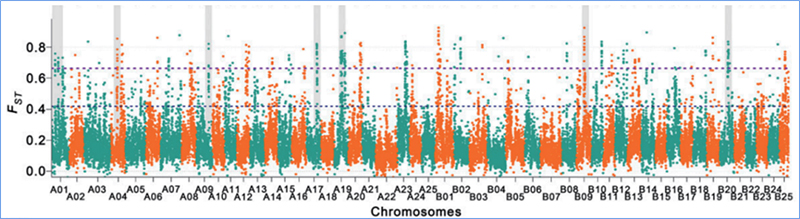
Wu, et. al.,Molekulare Pflanze, 2018
3.Genfluss
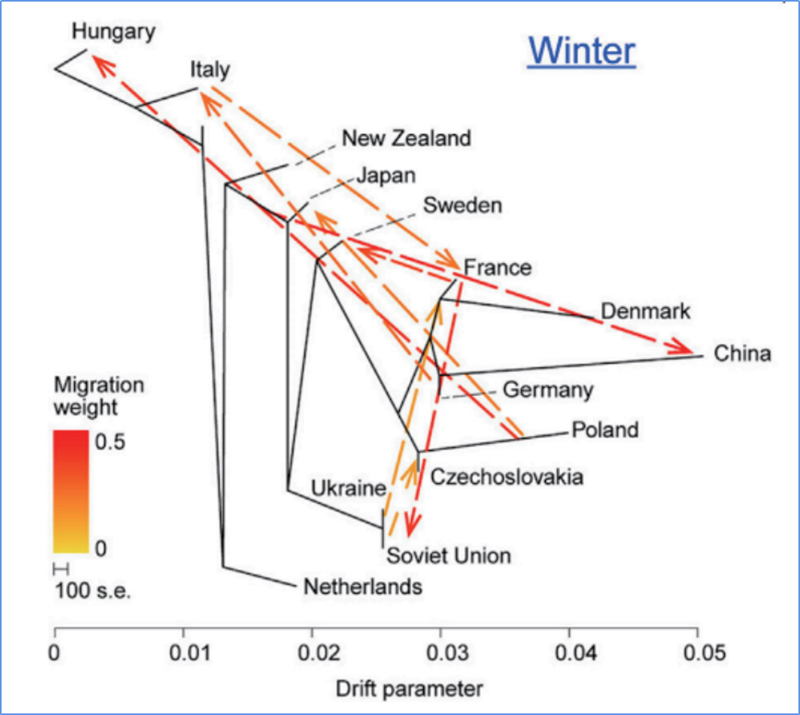
Wu, et. al.,Molekulare Pflanze, 2018
4.Demografische Geschichte
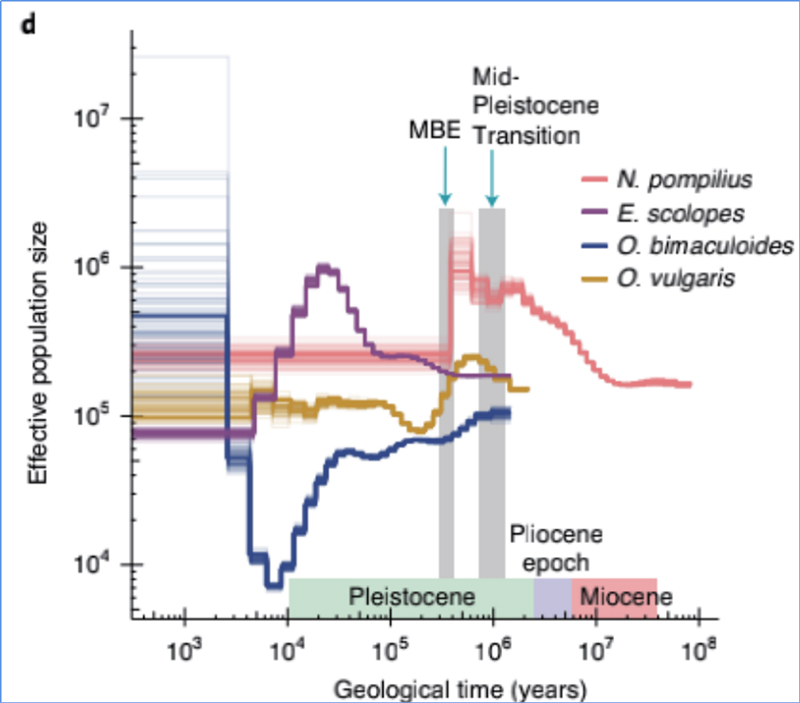
Zhang et al. al.,Naturökologie und Evolution, 2021
5.Divergenzzeit
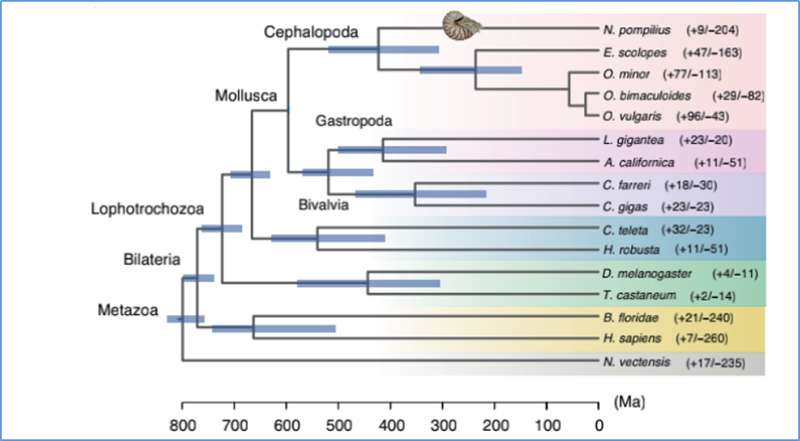
Zhang et al. al.,Naturökologie und Evolution, 2021
Entdecken Sie die Fortschritte, die durch die evolutionären Genetikdienste von BMKGene ermöglicht werden, anhand einer kuratierten Sammlung von Veröffentlichungen:
Hassanyar, AK et al. (2023) „Entdeckung molekularer SNP-Marker und Kandidatengene, die mit der Sackbrutvirus-Resistenz in Apis cerana cerana-Larven durch Gesamtgenom-Resequenzierung assoziiert sind“,Internationale Zeitschrift für Molekularwissenschaften, 24(7). doi: 10.3390/IJMS24076238.
Chai, J. et al. (2022) „Die Entdeckung eines wilden, genetisch reinen chinesischen Riesensalamanders schafft neue Möglichkeiten für den Naturschutz“,Zoologische Forschung, 2022, Bd. 43, Ausgabe 3, Seiten: 469–480, 43(3), S. 469–480. doi: 10.24272/J.ISSN.2095-8137.2022.101.
Han, M. et al. (2022) „Phylogeographisches Muster und Geschichte der Bevölkerungsentwicklung des indigenen Elymus sibiricus L. auf dem Qinghai-Tibetischen Plateau“,Grenzen der Pflanzenwissenschaft, 13, S. 882601. doi: 10.3389/FPLS.2022.882601/BIBTEX.
Wang, J. et al. (2022) „Genomische Einblicke in die Longan-Evolution aus einer Genomassemblierung auf Chromosomenebene und Populationsgenomik von Longan-Akzessionen“,Gartenbauforschung, 9. doi: 10.1093/HR/UHAC021.





