

Vergleichende Genomik

Servicevorteile
●Umfangreiche Expertise und Publikationsaufzeichnungen: Insgesamt hat BMKGene über 90 vergleichende Genomikprojekte abgeschlossen, mit einem kumulierten Impact Factor von 900.
●Umfassende bioinformatische Analyse: Das Analysepaket enthält die acht am häufigsten benötigten Analysen, liefert gut gestaltete, veröffentlichungsfertige Zahlen und ermöglicht eine einfache Interpretation der Ergebnisse
●Hochqualifiziertes Bioinformatik-Team und kurze Analysezyklen: Mit großer Erfahrung in der vergleichenden Genomanalyse erfüllt das Team von BMKGene vielfältige personalisierte Analyseanforderungen in kurzer Bearbeitungszeit
●Post-Sales-Support:Unser Engagement geht über den Projektabschluss hinaus und umfasst einen dreimonatigen Kundendienstzeitraum. Während dieser Zeit bieten wir Projektnachverfolgung, Unterstützung bei der Fehlerbehebung und Frage-und-Antwort-Sitzungen an, um alle Fragen zu den Ergebnissen zu beantworten.
Leistungsbeschreibung
| Geschätzte Bearbeitungszeit | Anzahl der Arten | Analysen |
| 30 Werktage | 6 - 12 | Clusterbildung von Genfamilien Erweiterung und Kontraktion der Genfamilie Phylogenetischer Baumaufbau Schätzung der Divergenzzeit (Fossile Kalibrierung erforderlich) LTR-Einfügezeit (für Pflanzen) Vervielfältigung des gesamten Genoms (für Pflanzen) Selektiver Druck Synteny-Analyse |
Bioinformatische Analysen
● Genfamilie
● Phylogenetik
● Divergenzzeit
● Selektiver Druck
● Synteny-Analyse
Musteranforderungen und Lieferung
Probenanforderungen:
Gewebe oder DNA zur Genomsequenzierung und -assemblierung
Für Gewebe
| Spezies | Gewebe | Umfrage | PacBio CCS |
| Tier | Viszerales Gewebe | 0,5 ~ 1 g | ≥ 3,5 g |
| Muskelgewebe | |||
| ≥ 5,0 g | |||
| ≥ 5,0 ml | |||
| Säugetierblut | |||
| ≥ 0,5 ml | |||
| Geflügel-/Fischblut | |||
| Anlage | Frisches Blatt | 1 ~ 2 g | ≥ 5,0 g |
| Blütenblatt/Stiel | 1 ~ 2 g | ≥ 10,0 g | |
| Wurzel/Samen | 1 ~ 2 g | ≥ 20,0 g | |
| Zellen | Kultivierte Zelle | - | ≥ 1 x 108 |
Daten
Genomsequenzdateien (.fasta) und Annotationsdateien (.gff3) eng verwandter Arten
Service-Workflow

Experimentdesign

Musterlieferung

Bibliotheksbau

Sequenzierung

Datenanalyse

Kundendienst
*Die hier gezeigten Demo-Ergebnisse stammen alle von Genomen, die mit Biomarker Technologies veröffentlicht wurden
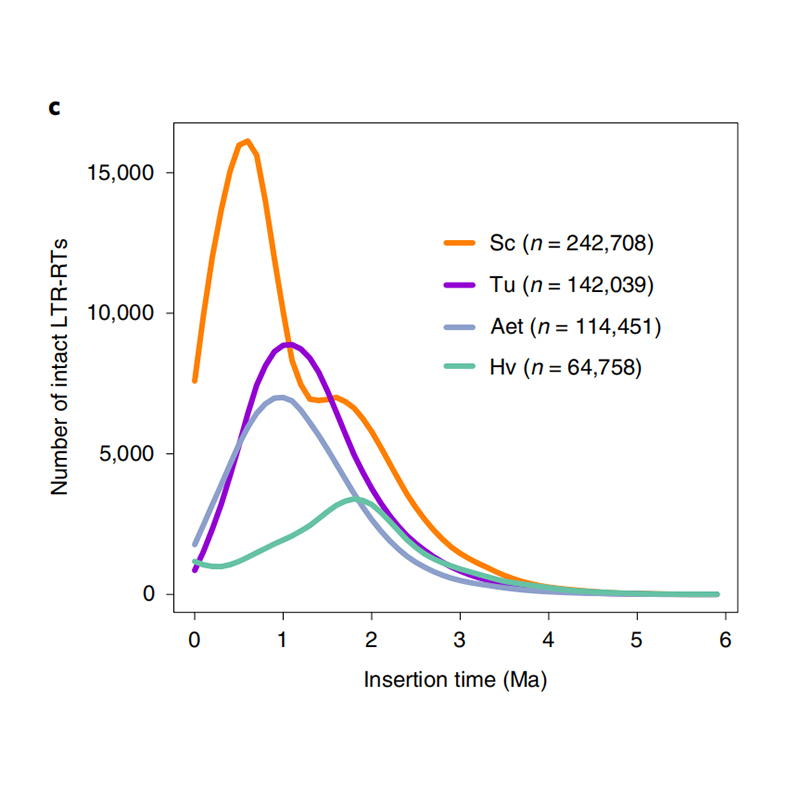
1. Schätzung der LTR-Insertionszeit: Die Abbildung zeigt eine einzigartige bimodale Verteilung der LTR-RTs-Insertionszeiten im Weining-Roggengenom im Vergleich zu anderen Arten. Der jüngste Höhepunkt trat vor etwa 0,5 Millionen Jahren auf.
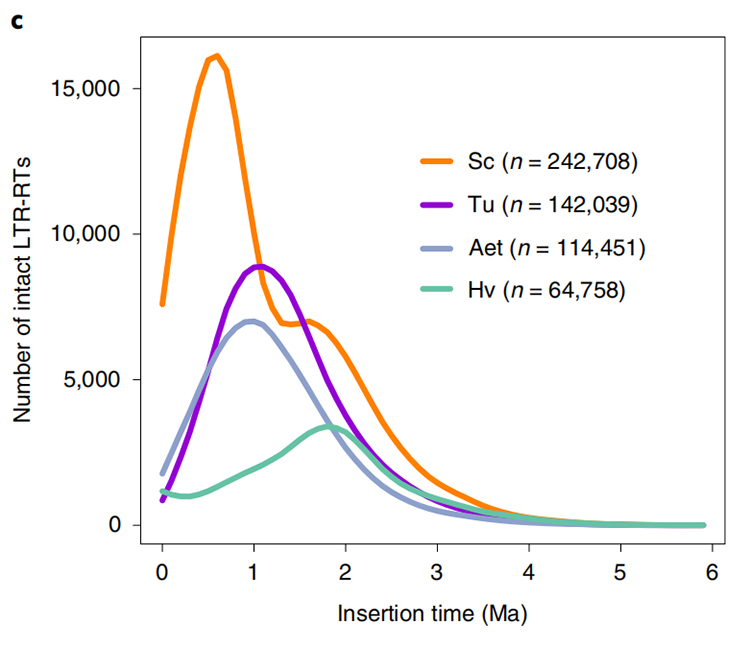
Li Guang et al.,Naturgenetik, 2021
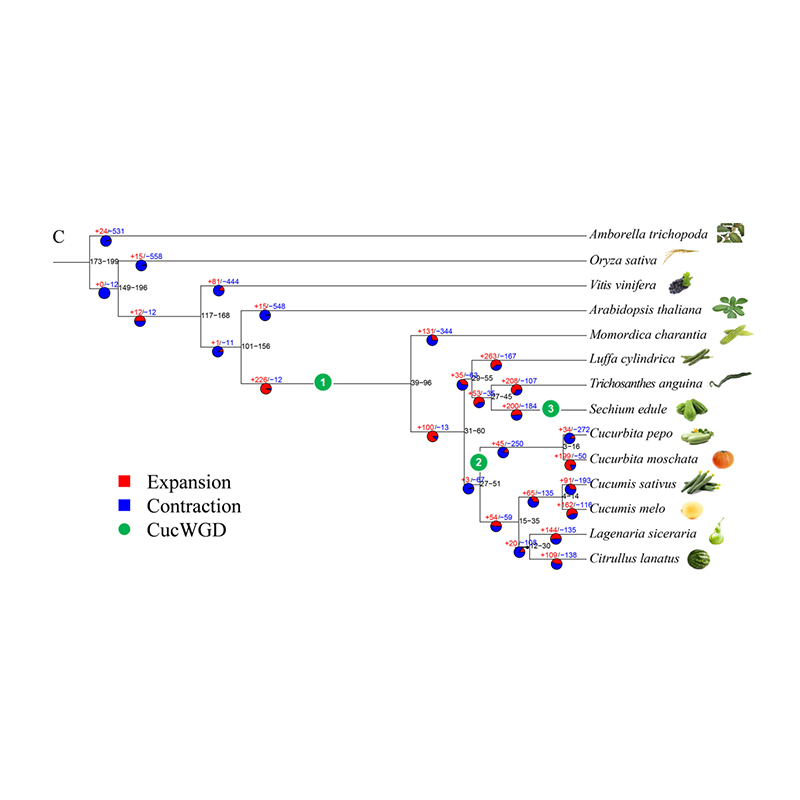
2. Phylogenie- und Genfamilienanalyse von Chayote (Sechium edule): Durch die Analyse von Chayote und den anderen 13 verwandten Arten in der Genfamilie wurde festgestellt, dass Chayote am engsten mit dem Schlangenkürbis (Trichosanthes anguina) verwandt ist. Chayote stammte aus Schlangenkürbis vor etwa 27–45 Millionen Jahren, und bei Chayote wurde vor 25 ± 4 Millionen Jahren eine Duplikation des gesamten Genoms (WGD) beobachtet, was das dritte WGD-Ereignis bei Cucuibitaceae darstellt.
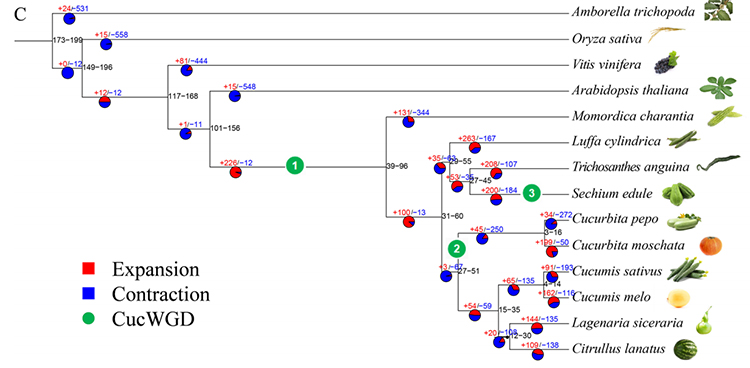
Fu A et al.,Gartenbauforschung, 2021
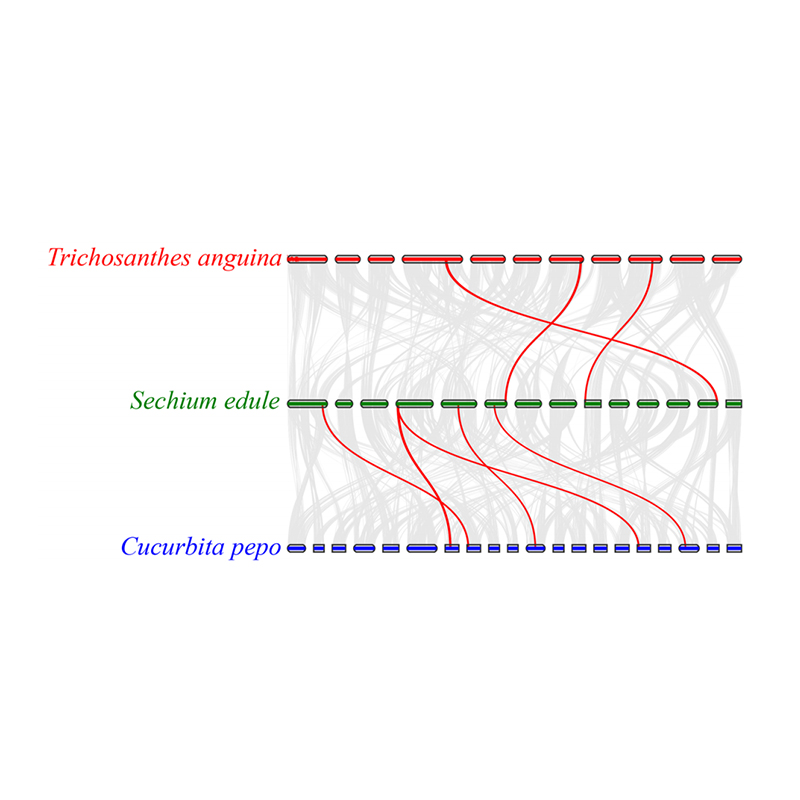
3. Synteny-Analyse: Einige Gene, die mit Phytohormonen bei der Fruchtentwicklung in Zusammenhang stehen, wurden in Chayote, Schlangenkürbis und Kürbis gefunden. Die Korrelation zwischen Chayote und Kürbis ist etwas höher als die zwischen Chayote und Schlangenkürbis.
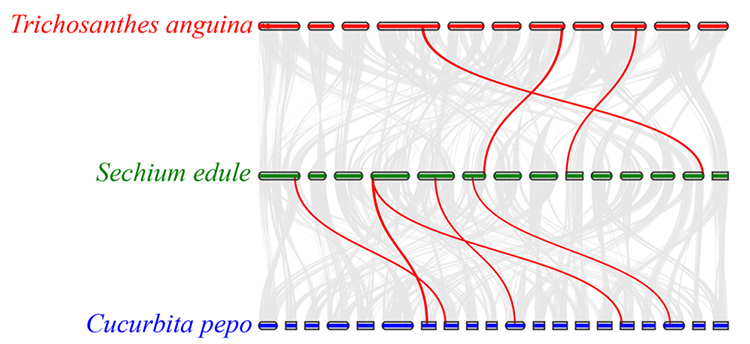
Fu A et al.,Gartenbauforschung, 2021
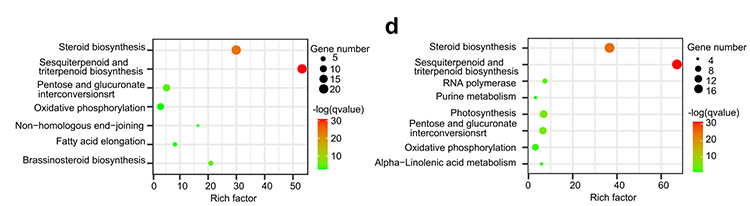
4.Genfamilienanalyse: Die KEGG-Anreicherung bei der Genfamilienexpansion und -kontraktion in den Genomen von G.thurberi und G.davidsonii zeigte, dass Gene im Zusammenhang mit der Steroidbiosynthese und der Brassinosteroidbiosynthese erweitert wurden.
Yang Z et al.,BMC-Biologie, 2021
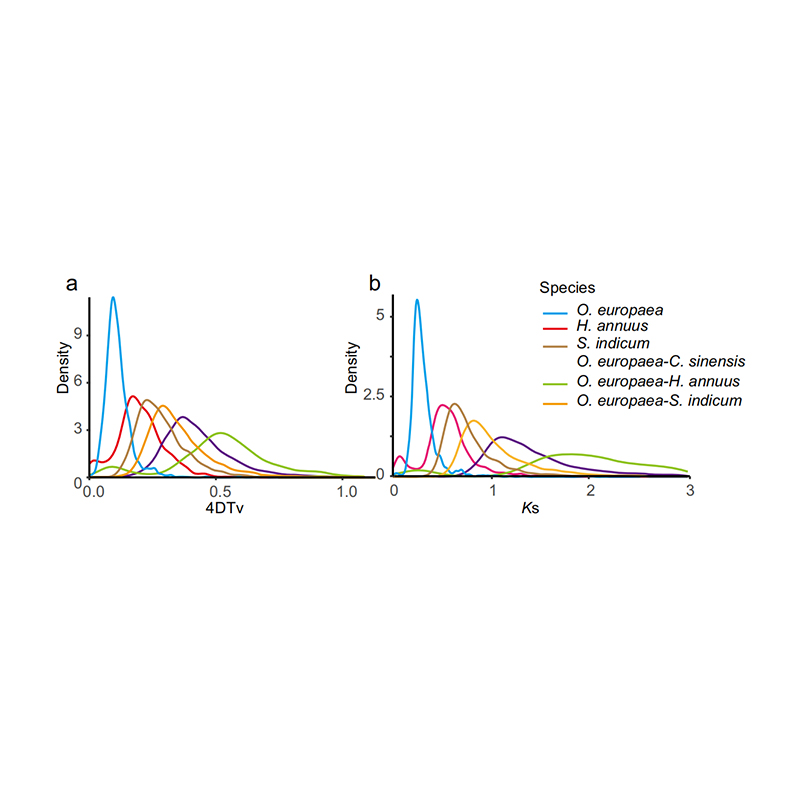
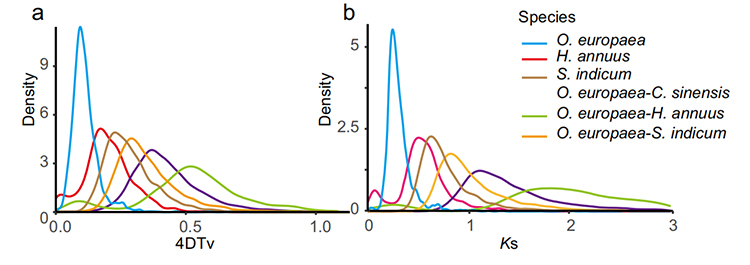
5. Analyse der Duplikation des gesamten Genoms: Die 4DTV- und Ks-Verteilungsanalyse zeigte das Duplikationsereignis des gesamten Genoms. Spitzenwerte von Intraspezies zeigten Duplikationsereignisse. Spitzenwerte von Interspezies-Artbildungsereignissen. Die Analyse ergab, dass O. europaea im Vergleich zu den anderen drei eng verwandten Arten in jüngerer Zeit eine groß angelegte Genduplikation durchlief.
Rao G et al.,Gartenbauforschung, 2021
BMK-Fall
Rose ohne Stachel: genomische Erkenntnisse im Zusammenhang mit der Feuchtigkeitsanpassung
Veröffentlicht: National Science Review, 2021
Sequenzierungsstrategie:
'Basye'sDornenlos' (R.Wichurainan) Genom:
Ca. 93 X PacBio + ca. 90 x Nanopore + 267 x Illumina
Wichtigste Ergebnisse
1. Hochwertiges R.wichuraiana-Genom wurde mithilfe von Long-Read-Sequenzierungstechniken konstruiert, die eine Gesamtheit von 530,07 MB ergeben (die geschätzte Genomgröße betrug laut Durchflusszytometrie etwa 525,9 MB und laut Genomuntersuchung 525,5 MB; die Heterozygotie betrug etwa 1,03 %). Der von BUSCO geschätzte Wert lag bei 93,9 %. Im Vergleich zu „Old Blush“ (haploOB) wurde die Qualität und Vollständigkeit dieses Genoms durch die Basis-Single-Base-Genauigkeit und den LTR-Assembly-Index (LAI = 20,03) bestätigt. Das Genom von R.wichuraiana enthält 32.674 proteinkodierende Gene.
2. Die gemeinsame Multi-Omics-Analyse, bestehend aus vergleichender Genomik, Transkriptomik und QTL-Analyse der genetischen Population, enthüllte die entscheidende Artbildung zwischen R. wichuraiana und Rosa chinensis. Außerdem war es wahrscheinlich, dass die Variation der Expression verwandter Gene in QTL mit der Strukturierung von Stammstachelmustern zusammenhängt.
Eine vergleichende genomische Analyse zwischen Basye's Thornless und Rosa chinensis, einschließlich Syntenieanalyse, Genfamiliencluster sowie Expansions- und Kontraktionsanalyse, ergab eine große Anzahl von Variationen, die sich auf entscheidende Merkmale bei Rosen beziehen. Die einzigartige Erweiterung der NAC- und FAR1/FRS-Genfamilie war höchstwahrscheinlich mit einer Resistenz gegen Black Spot verbunden.
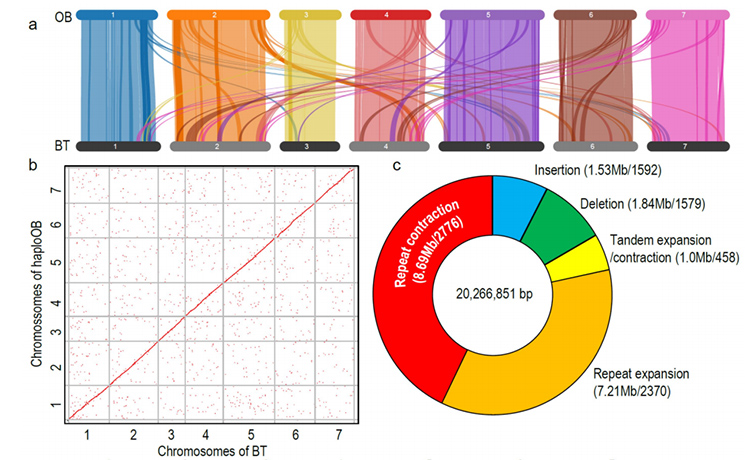

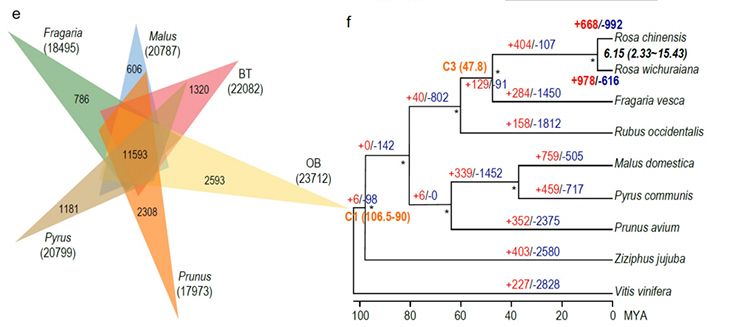
Vergleichende Genomanalyse zwischen BT- und haploOB-Genomen.
Zhong, M., et al. „Rose ohne Stachel: genomische Erkenntnisse im Zusammenhang mit Feuchtigkeitsanpassung“National Science Review, 2021;, nwab092.





