

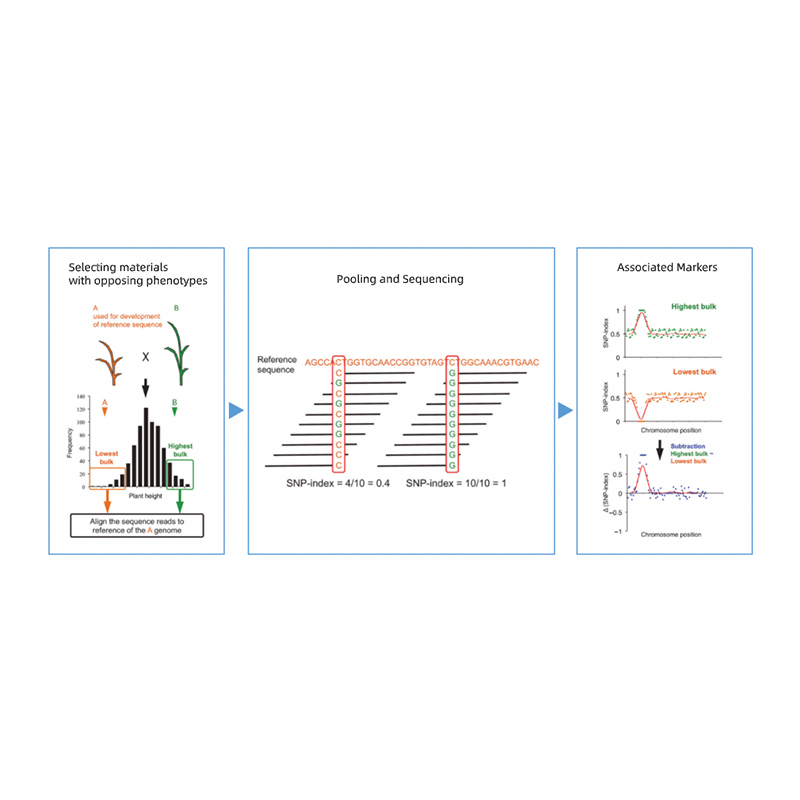
Bulled Segregant Analysis

Servicevorteile
Takagi et al., The Plant Journal, 2013
● Genauige Lokalisierung: Mischen von Massen mit 30+30 bis 200+200 Personen, um Hintergrundgeräusche zu minimieren; Nichtsynonyme Mutatantbasis-Kandidatenregion Vorhersage.
● Umfassende Analyse: Annotation für eingehende Kandidatengenfunktion, einschließlich NR, Swissprot, Go, Kegg, Cog, KOG usw.
● Schnellere Turnaround -Zeit: Schnelle Genlokalisierung innerhalb von 45 Arbeitstagen.
● Umfangreiche Erfahrung: BMK hat in Tausenden von Merkmalen Lokalisierung beigetragen, die verschiedene Arten wie Pflanzen, Wasserprodukte, Wald, Blumen, Früchte usw. abdecken.
Servicespezifikationen
Bevölkerung:
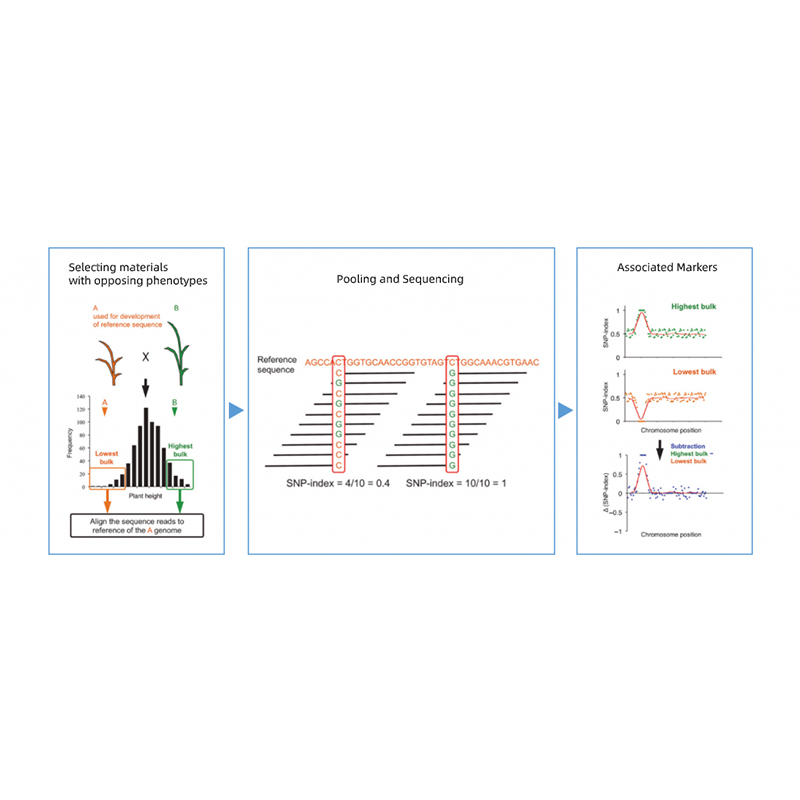
Trennung von Nachkommen von Eltern mit gegnerischen Phänotypen.
EG F2 Nachkommen, Backcross (BC), rekombinante Inzuchtlinie (RIL)
Mischpool
Für qualitative Merkmale: 30 bis 50 Personen (mindestens 20)/Masse
Für quantitative Tratis: Top 5% bis 10% Personen mit beiden extremen Phänotypen in der gesamten Bevölkerung (mindestens 30+30).
Empfohlene Sequenzierungstiefe
Mindestens 20x/übergeordnet und 1x/Nachkommen -Individuum (z.
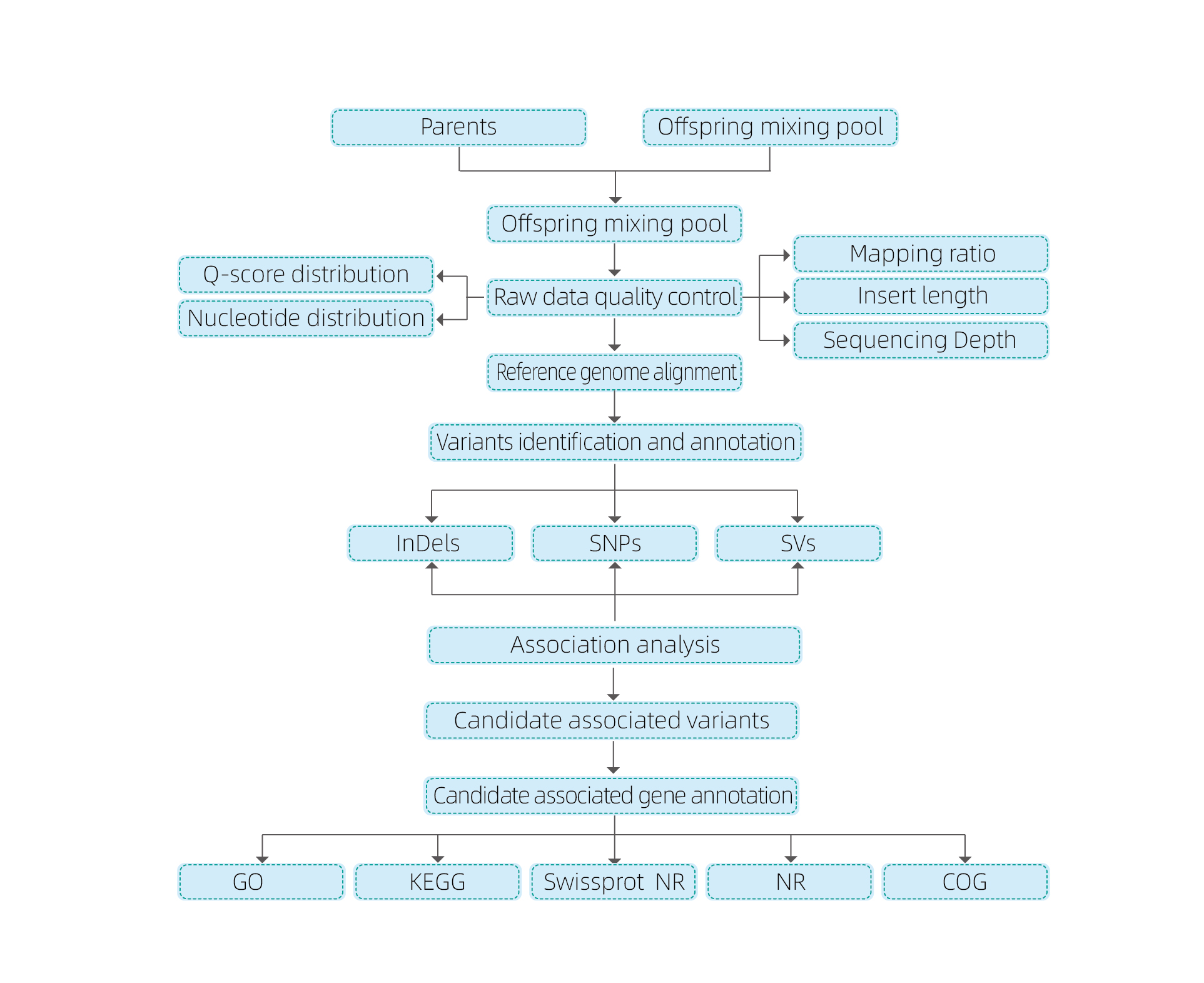
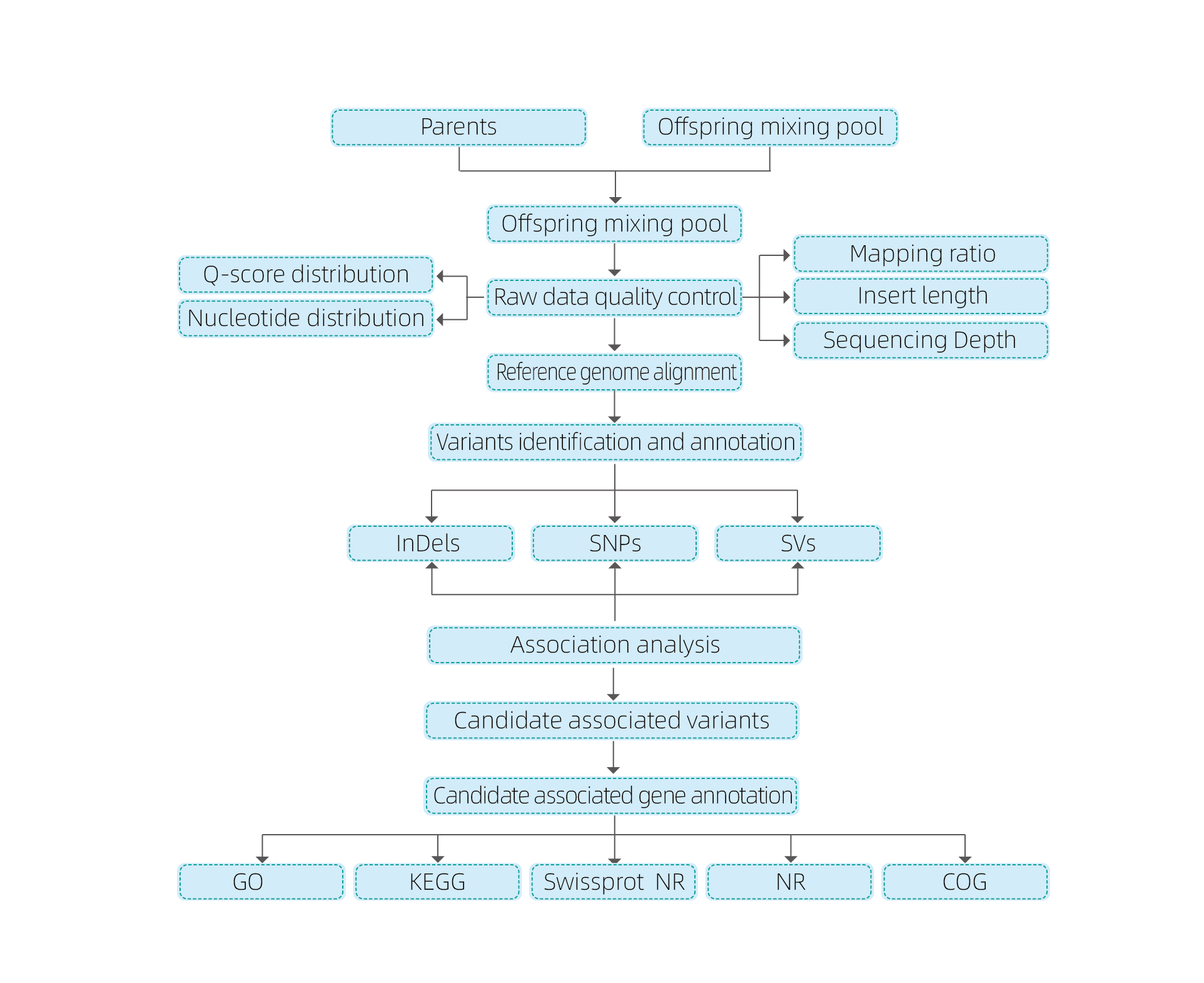
Bioinformatikanalysen
● Das gesamte Genom -Neuausgleich
● Datenverarbeitung
● SNP/Indel Calling
● Screening der Kandidatenregion
● Annotation der Genefunktionskandidat
Probenanforderungen und Lieferung
Beispielanforderungen:
Nukleotide:
| GDNA -Probe | Gewebeprobe |
| Konzentration: ≥ 30 ng/μl | Pflanzen: 1-2 g |
| Menge: ≥2 μg (FURMB ≥ 15 μl) | Tiere: 0,5-1 g |
| Reinheit: OD260/280 = 1,6-2,5 | Vollblut: 1,5 ml |
Service Work Flow

Experimententwurf

Probenabgabe

RNA -Extraktion

Bibliothekskonstruktion

Sequenzierung

Datenanalyse

Nachverkaufsdienste
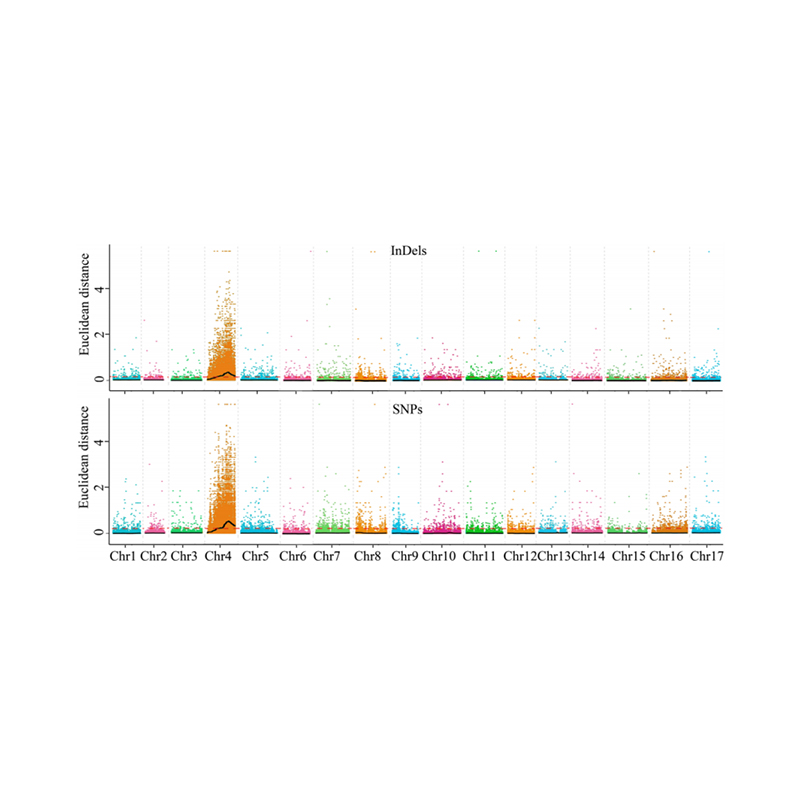
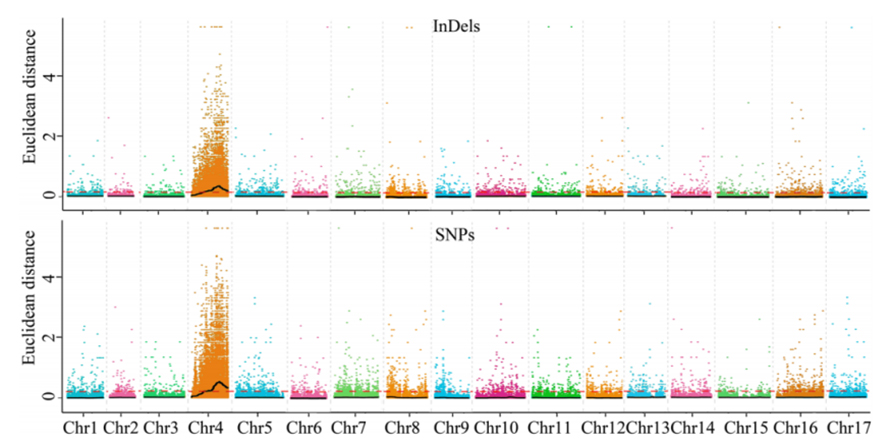
1. Assoziationsanalyse Basis auf euklidischer Distanz (ED), um die Kandidatenregion zu identifizieren. In der folgenden Abbildung
X-Achse: Chromosomenzahl; Jeder Punkt stellt einen ED -Wert eines SNP dar. Die schwarze Linie entspricht dem ausgestatteten ED -Wert. Ein höherer ED -Wert zeigt einen signifikanteren Zusammenhang zwischen der Stelle und dem Phänotyp an. Die rote Dash -Linie repräsentiert den Schwellenwert einer signifikanten Assoziation.
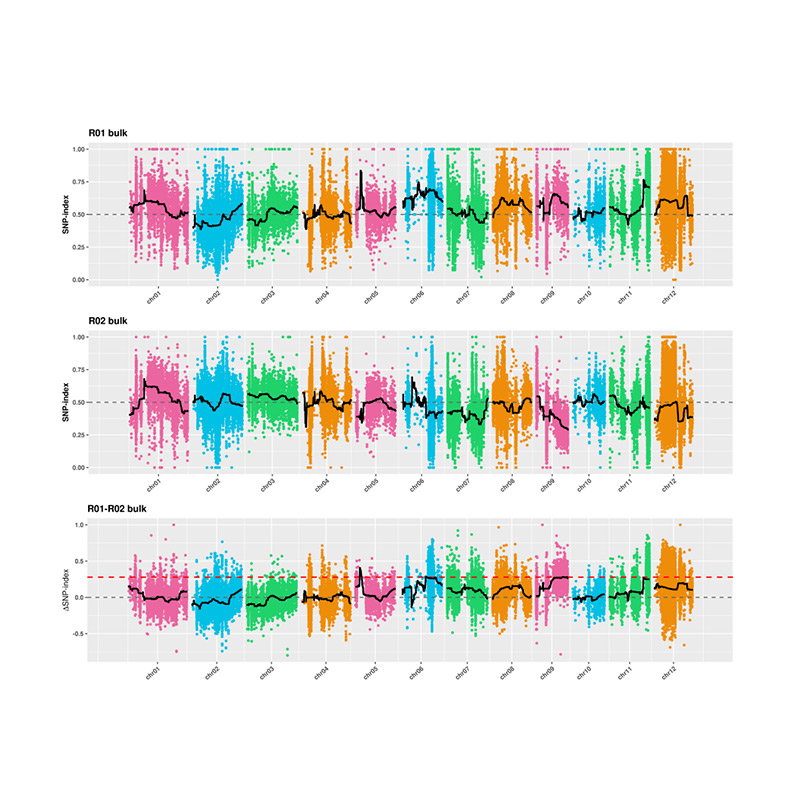
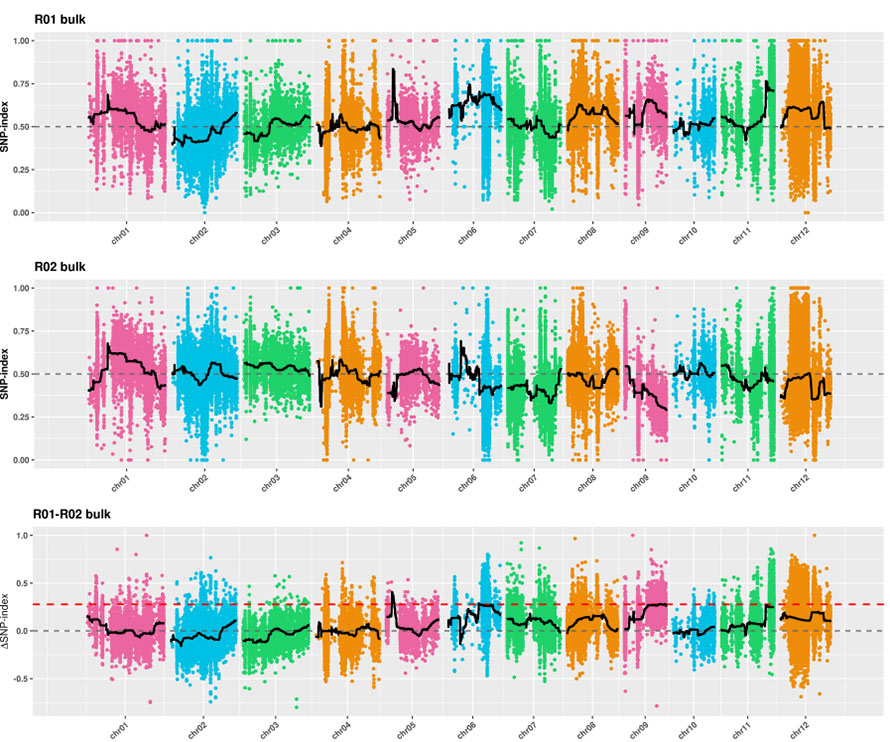
2. Assoziationsanalyse basiert kein SNP-Index
X-Achse: Chromosomenzahl; Jeder Punkt repräsentiert den SNP-Index-Wert. Die schwarze Linie steht für einen angepassten SNP-Index-Wert. Je größer der Wert ist, desto bedeutender ist der Assoziation.
BMK -Fall
Der quantitative Merkmal des Haupteffekts fnnl7.1 codiert ein vorhandenes Protein der späten Embryogenese, die mit der Länge von Fruchthals in der Gurke assoziiert sind
Veröffentlicht: Plant Biotechnology Journal, 2020
Sequenzierungsstrategie:
Eltern (JIN5-508, YN): Das gesamte Genom-Neuausgleich für 34 × und 20 ×.
DNA-Pools (50 Langhals und 50 Kurzgehe): Wiederholung für 61 × und 52 ×
Schlüsselergebnisse
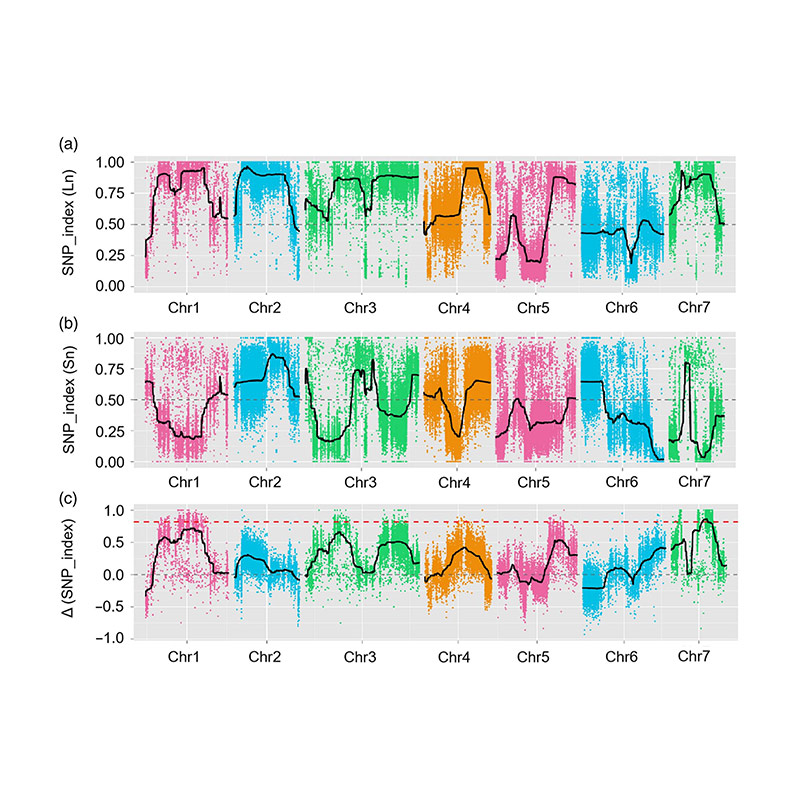
In dieser Studie wurde die segregierte Bevölkerung (F2 und F2: 3) durch Überqueren der Long-Neck-Gurkenlinie JIN5-508 und kurzhals YN erzeugt. Zwei DNA-Pools wurden von 50 extremen Langzeitpersonen und 50 extremen Kurznack-Personen konstruiert. Die Haupteffekt-QTL wurde auf CHR07 durch BSA-Analyse und herkömmliches QTL-Mapping identifiziert. Die Kandidatenregion wurde durch Feindmaking, Genexpressionsquantifizierung und transgene Experimente weiter eingeengt, die ein Schlüsselgen bei der Kontrolle der Nackenlänge, CSFNL7.1, enthüllten. Darüber hinaus wurde festgestellt, dass Polymorphismus in der CSFNL7.1 -Promotorregion mit der entsprechenden Expression verbunden ist. Eine weitere phylogenetische Analyse deutete darauf hin, dass der FNL7.1 -Ort sehr wahrscheinlich aus Indien stammt.
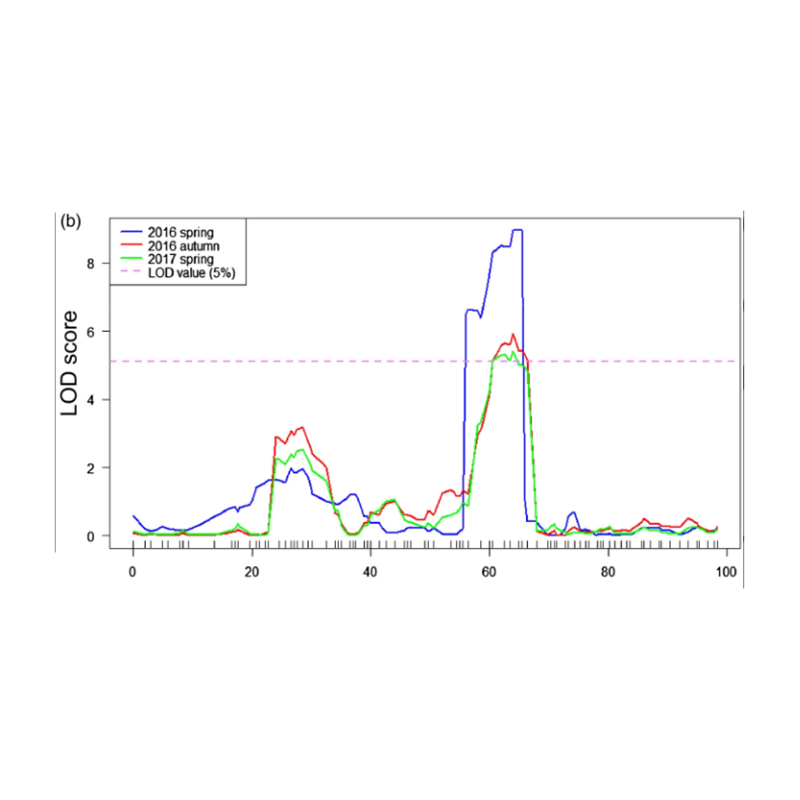
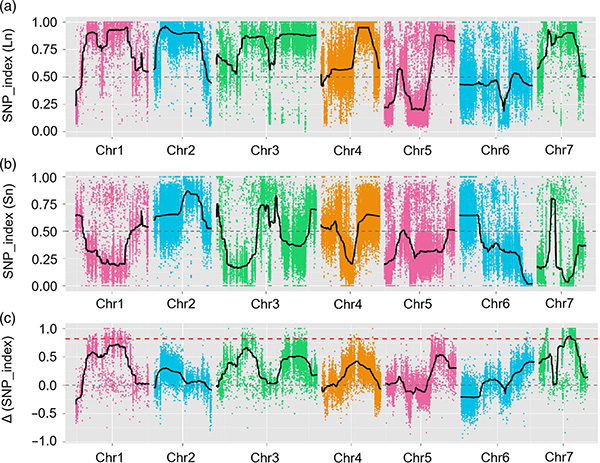 QTL-Mapping in der BSA | 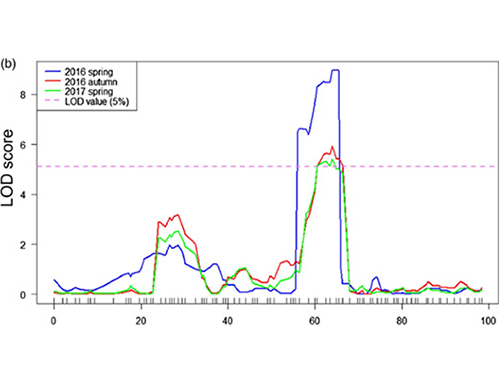 LOD-Profile von QTL-Länge von Gurken-Nackenlänge, die auf CHR07 identifiziert wurden |
Xu, X., et al. "Der quantitative Merkmal des Hauptseffekts fnl7.1 codiert ein vorhandenes Protein der späten Embryogenese, das mit der Länge der Fruchthals in der Gurke assoziiert ist." Plant Biotechnology Journal 18.7 (2020).



