

BMKMANU S1000 räumliches Transkriptom

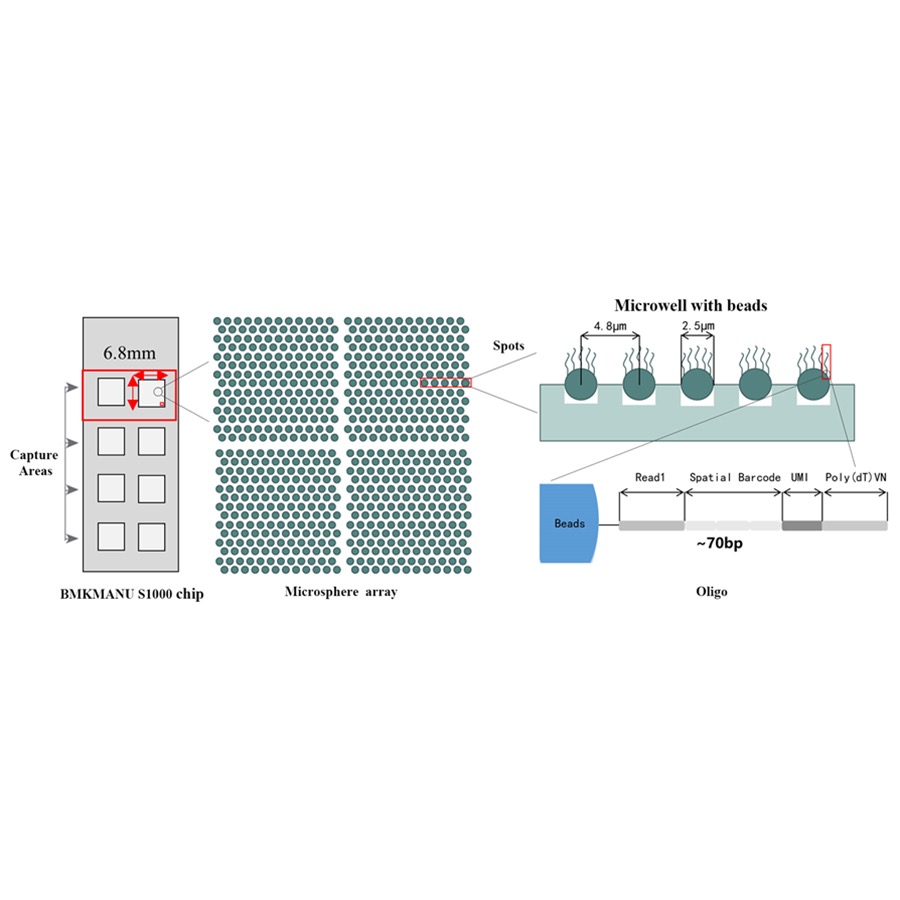
Technisches Schema des räumlichen Transkriptoms BMKMANU S1000

Merkmale
● Auflösung: 5 µM
● Punktdurchmesser: 2,5 µM
● Anzahl der Spots: ca. 2 Millionen
● 3 mögliche Aufnahmebereichsformate: 6,8 mm * 6,8 mm, 11 mm * 11 mm oder 15 mm * 20 mm
● Jede Perle mit Barcode ist mit Primern beladen, die aus 4 Abschnitten bestehen:
Poly(dT)-Schwanz für mRNA-Priming und cDNA-Synthese
Einzigartiger molekularer Identifikator (UMI) zur Korrektur von Amplifikationsverzerrungen
Räumlicher Barcode
Bindungssequenz des Teil-Read-1-Sequenzierungsprimers
● H&E und Fluoreszenzfärbung von Schnitten
● Möglichkeit zur NutzungZellsegmentierungstechnologie: Integration von H&E-Färbung, Fluoreszenzfärbung und RNA-Sequenzierung, um die Grenzen jeder Zelle zu bestimmen und die Genexpression jeder Zelle korrekt zuzuordnen.
Vorteile von BMKMANU S1000
●Subzelluläre Auflösung: Jeder Erfassungsbereich enthielt >2 Millionen räumliche Barcode-Spots mit einem Durchmesser von 2,5 µm und einem Abstand von 5 µm zwischen den Spotzentren, was eine räumliche Transkriptomanalyse mit subzellulärer Auflösung (5 µm) ermöglichte.

●Mehrstufige Auflösungsanalyse:Flexible mehrstufige Analyse im Bereich von 100 μm bis 5 μm zur Auflösung verschiedener Gewebemerkmale mit optimaler Auflösung.

●Möglichkeit zur Verwendung der „Drei-in-einem-Folie“-Zellsegmentierungstechnologie:Durch die Kombination von Fluoreszenzfärbung, H&E-Färbung und RNA-Sequenzierung auf einem einzigen Objektträger ermöglicht unser „Drei-in-eins“-Analysealgorithmus die Identifizierung von Zellgrenzen für die anschließende zellbasierte Transkriptomik.

●Kompatibel mit mehreren Sequenzierungsplattformen: Sowohl NGS- als auch Long-Read-Sequenzierung verfügbar.
●Flexible Gestaltung von 1–8 aktiven Erfassungsbereichen: Die Größe des Aufnahmebereichs ist flexibel und ermöglicht die Verwendung von 3 Formaten (6,8 mm * 6,8 mm, 11 mm * 11 mm und 15 mm * 20 mm).
●Service aus einer Hand: Es integriert alle erfahrungs- und kompetenzbasierten Schritte, einschließlich Kryoschnitte, Färbung, Gewebeoptimierung, räumliche Barcodes, Bibliotheksvorbereitung, Sequenzierung und Bioinformatik.
●Umfassende Bioinformatik und benutzerfreundliche Visualisierung der Ergebnisse:Das Paket umfasst 29 Analysen und über 100 hochwertige Abbildungen, kombiniert mit der Verwendung selbst entwickelter Software zur Visualisierung und Anpassung der Zellteilung und Spot-Clusterbildung.
●Maßgeschneiderte Datenanalyse und Visualisierung: verfügbar für verschiedene Forschungsanfragen
●Hochqualifiziertes technisches Team: mit Erfahrung in über 250 Gewebetypen und über 100 Arten, darunter Menschen, Mäuse, Säugetiere, Fische und Pflanzen.
●Echtzeit-Updates für das gesamte Projekt: mit voller Kontrolle über den experimentellen Fortschritt.
●Optionale gemeinsame Analyse mit Einzelzell-mRNA-Sequenzierung
Leistungsbeschreibung
|
Probe Anforderungen
| Bibliothek |
Sequenzierungsstrategie
| Daten empfohlen | Qualitätskontrolle |
| OCT-eingebettete Kryoproben, 3 Blöcke pro Probe | S1000-cDNA-Bibliothek | Illumina PE150 (andere Plattformen verfügbar) | 100.000 PE-Lesevorgänge pro 100 µM (60-150 GB) | RIN>7 |
Für weitere Informationen zur Anleitung zur Probenvorbereitung und zum Service-Workflow wenden Sie sich bitte an aBMKGENE-Experte
Service-Workflow
In der Probenvorbereitungsphase wird zunächst ein Massen-RNA-Extraktionsversuch durchgeführt, um sicherzustellen, dass eine qualitativ hochwertige RNA erhalten werden kann. In der Gewebeoptimierungsphase werden die Schnitte gefärbt und sichtbar gemacht und die Permeabilisierungsbedingungen für die mRNA-Freisetzung aus dem Gewebe optimiert. Das optimierte Protokoll wird dann beim Bibliotheksaufbau angewendet, gefolgt von der Sequenzierung und Datenanalyse.
Der gesamte Service-Workflow umfasst Echtzeitaktualisierungen und Kundenbestätigungen, um eine reaktionsfähige Feedbackschleife aufrechtzuerhalten und eine reibungslose Projektabwicklung sicherzustellen.


Die von BMKMANU S1000 generierten Daten werden mit der von BMKGENE unabhängig entwickelten Software „BSTMatrix“ analysiert und eine Genexpressionsmatrix erstellt. Von dort aus wird ein Standardbericht erstellt, der die Datenqualitätskontrolle, die Analyse innerhalb der Stichprobe und die Analyse zwischen den Gruppen umfasst.
● Datenqualitätskontrolle:
Datenausgabe und Verteilung des Qualitätsfaktors
Generkennung pro Spot
Gewebeabdeckung
● Inner-Sample-Analyse:
Genreichtum
Spot-Clustering, einschließlich Analyse reduzierter Dimensionen
Differenzielle Expressionsanalyse zwischen Clustern: Identifizierung von Markergenen
Funktionelle Annotation und Anreicherung von Markergenen
● Intergruppenanalyse:
Rekombination von Spots aus beiden Proben (z. B. erkrankte und Kontrollproben) und erneutes Clustern
Identifizierung von Markergenen für jeden Cluster
Funktionelle Annotation und Anreicherung von Markergenen
Differentialausdruck desselben Clusters zwischen Gruppen
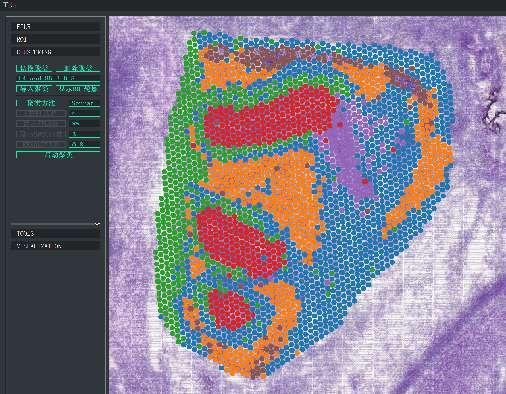
Darüber hinaus hat BMKGENE „BSTViewer“ entwickelt, ein benutzerfreundliches Tool, das es dem Benutzer ermöglicht, die Genexpression und Spot-Clustering in verschiedenen Auflösungen zu visualisieren.
BMKGene hat eine Software zur benutzerfreundlichen Visualisierung entwickelt
BSTViewer-Spot-Clustering mit mehrstufiger Auflösung

BSTCellViewer: automatische und manuelle Zellaufteilung

Inner-Sample-Analyse
Spot-Clustering:

Identifizierung und räumliche Verteilung von Markergenen:

Intergruppenanalyse
Datenkombination aus beiden Gruppen und Re-Cluster:

Markergene neuer Cluster:

Entdecken Sie die Fortschritte, die durch die räumlichen Transkriptomikdienste von BMKGene mit der BMKManu S1000-Technologie in dieser vorgestellten Veröffentlichung ermöglicht werden:
Song, X. et al. (2023) „Räumliche Transkriptomik zeigt lichtinduzierte Chlorenchymzellen, die an der Förderung der Triebregeneration im Tomatenkallus beteiligt sind“,Tagungsband der National Academy of Sciences der Vereinigten Staaten von Amerika, 120(38), S. e2310163120. doi: 10.1073/pnas.2310163120
Sie, Y. et al. (2023) „Systematischer Vergleich sequenzierungsbasierter räumlicher Transkriptommethoden“,bioRxiv, P. 2023.12.03.569744. doi: 10.1101/2023.12.03.569744.






{kind=link}