

Prostorový přepis BMKMANU S1000

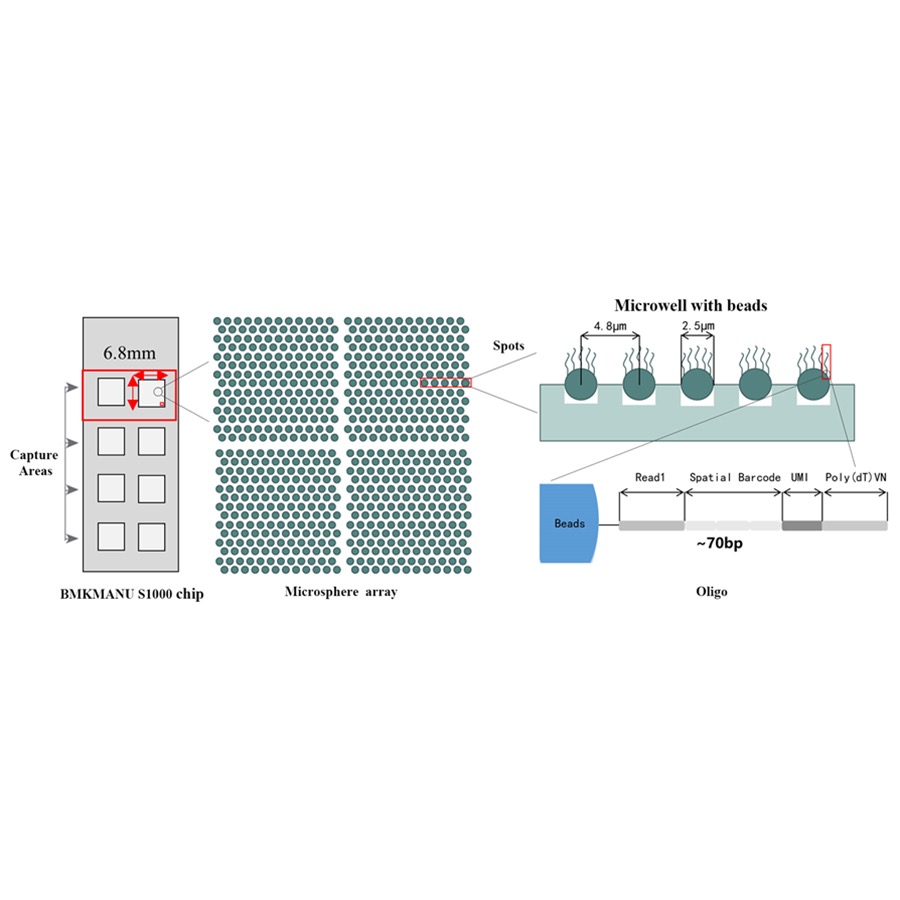
Technické schéma prostorového transkriptomu BMKMANU S1000

Vlastnosti
● Rozlišení: 5 µM
● Průměr bodu: 2,5 µM
● Počet spotů: přibližně 2 miliony
● 3 možné formáty snímané oblasti: 6,8 mm * 6,8 mm, 11 mm * 11 mm nebo 15 mm * 20 mm
● Každá kulička s čárovým kódem je naplněna primery složenými ze 4 částí:
poly(dT) konec pro priming mRNA a syntézu cDNA
Unikátní molekulární identifikátor (UMI) pro korekci zkreslení amplifikace
Prostorový čárový kód
Vazebná sekvence sekvenačního primeru částečného čtení 1
● H&E a fluorescenční barvení řezů
● Možnost použitítechnologie buněčné segmentace: integrace H&E barvení, fluorescenčního barvení a sekvenování RNA pro určení hranic každé buňky a správné přiřazení genové exprese každé buňce.
Výhody BMKMANU S1000
●Subcelulární rozlišení: Každá zachycená oblast obsahovala >2 miliony prostorových čárových kódů o průměru 2,5 µm a rozestupem 5 µm mezi středy skvrn, což umožňuje analýzu prostorového transkriptomu se subcelulárním rozlišením (5 µm).

●Víceúrovňová analýza rozlišení:Flexibilní víceúrovňová analýza v rozsahu od 100 μm do 5 μm pro rozlišení různých tkáňových vlastností při optimálním rozlišení.

●Možnost použití technologie segmentace buněk „Tři v jednom snímku“:Kombinací fluorescenčního barvení, H&E barvení a sekvenování RNA na jednom sklíčku umožňuje náš analytický algoritmus „tři v jednom“ identifikaci buněčných hranic pro následnou transkriptomiku založenou na buňkách.

●Kompatibilní s vícenásobnými sekvenčními platformami: K dispozici jak NGS, tak dlouhé čtení sekvence.
●Flexibilní design 1-8 aktivních snímacích oblastí: Velikost snímací oblasti je flexibilní, je možné použít 3 formáty (6,8 mm * 6,8 mm., 11 mm * 11 mm a 15 mm * 20 mm)
●Jednorázová služba: Integruje všechny kroky založené na zkušenostech a dovednostech, včetně kryořezů, barvení, optimalizace tkání, prostorového čárového kódování, přípravy knihovny, sekvenování a bioinformatiky.
●Komplexní bioinformatika a uživatelsky přívětivá vizualizace výsledků:Balíček obsahuje 29 analýz a více než 100 vysoce kvalitních čísel v kombinaci s použitím vlastního softwaru vyvinutého pro vizualizaci a přizpůsobení dělení buněk a shlukování bodů.
●Vlastní analýza a vizualizace dat: k dispozici pro různé výzkumné požadavky
●Vysoce kvalifikovaný technický tým: se zkušenostmi s více než 250 typy tkání a více než 100 druhy včetně lidí, myší, savců, ryb a rostlin.
●Aktualizace celého projektu v reálném čase: s plnou kontrolou průběhu experimentu.
●Volitelná společná analýza s jednobuněčným sekvenováním mRNA
Specifikace služby
|
Ochutnat Požadavky
| Knihovna |
Strategie sekvenování
| Doporučené údaje | Kontrola kvality |
| Kryo vzorky vložené pomocí OCT, 3 bloky na vzorek | Knihovna cDNA S1000 | Illumina PE150 (k dispozici další platformy) | 100K PE čtení na 100 uM (60-150 Gb) | RIN>7 |
Chcete-li získat další podrobnosti o pokynech pro přípravu vzorků a pracovním postupu služeb, neváhejte se obrátit na aExpert BMKGENE
Tok servisní práce
Ve fázi přípravy vzorku je provedena počáteční zkouška hromadné extrakce RNA, aby bylo zajištěno, že lze získat vysoce kvalitní RNA. Ve fázi optimalizace tkáně jsou řezy obarveny a vizualizovány a jsou optimalizovány podmínky permeabilizace pro uvolňování mRNA z tkáně. Optimalizovaný protokol je poté aplikován při konstrukci knihovny, po které následuje sekvenování a analýza dat.
Kompletní pracovní postup služeb zahrnuje aktualizace v reálném čase a potvrzování klientů, aby byla zachována odezva zpětné vazby, což zajišťuje hladké provádění projektu.


Data generovaná BMKMANU S1000 jsou analyzována pomocí softwaru „BSTMatrix“, který je nezávisle navržen společností BMKGENE a generuje Gene Expression Matrix. Odtud se generuje standardní zpráva, která zahrnuje kontrolu kvality dat, analýzu vnitřních vzorků a analýzu mezi skupinami.
● Kontrola kvality dat:
Výstup dat a distribuce skóre kvality
Detekce genů na místě
Pokrytí tkání
● Analýza vnitřního vzorku:
Genové bohatství
Bodové shlukování, včetně analýzy redukovaných rozměrů
Diferenciální expresní analýza mezi shluky: identifikace markerových genů
Funkční anotace a obohacení markerových genů
● Meziskupinová analýza:
Rekombinace skvrn z obou vzorků (např. nemocných a kontrolních) a reshlukování
Identifikace markerových genů pro každý shluk
Funkční anotace a obohacení markerových genů
Diferenciální vyjádření stejného shluku mezi skupinami
Navíc BMKGENE vyvinul „BSTViewer“ je uživatelsky přívětivý nástroj, který umožňuje uživateli vizualizovat genovou expresi a shlukování bodů v různých rozlišeních.
BMKGene vyvinul software pro uživatelsky přívětivou vizualizaci
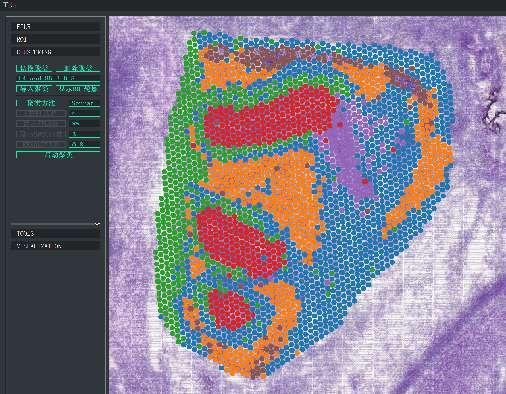
Bodové shlukování BSTViewer ve víceúrovňovém rozlišení

BSTCellViewer: automatické a manuální dělení buněk

Analýza vnitřního vzorku
Shlukování skvrn:

Identifikace markerových genů a prostorová distribuce:

Meziskupinová analýza
Kombinace dat z obou skupin a přeskupení:

Markerové geny nových klastrů:

V této doporučené publikaci prozkoumejte pokroky, které umožňují služby prostorové transkriptomiky BMKGene s technologií BMKManu S1000:
Song, X. a kol. (2023) „Prostorová transkriptomika odhaluje světlem indukované buňky chlorenchymu, které se podílejí na podpoře regenerace výhonků v kalusu rajčat“,Proceedings of the National Academy of Sciences of the United States of America, 120(38), str. e2310163120. doi: 10.1073/pnas.2310163120
Vy, Y. a spol. (2023) 'Systematické srovnávání prostorových transkriptomických metod založených na sekvenování',bioRxiv, str. 2023.12.03.569744. doi: 10.1101/2023.12.03.569744.






{kind=link}